


- 假设大家用GEM-X产生了一个测序文库,然后送去测序,测序送回来之后,我们通常会拿到我们的测序原始数据,比如fastq文件
- 这个时候,我们可以使用10x开发的Cell Ranger,去预处理原始的测序Reads,包括去提取细胞的信息,计算每个细胞中,每个基因的表达量,最终会获得一系列的Cell Ranger产生的文件
- 在Cell Ranger产出的诸多文件中,有一个文件的后缀名叫cloupe,这个cloupe文件可以直接在Loupe Browser中打开
- Loupe Browser是10x研发的,免费的桌面工具,可以运行在Windows和MacOS上,目前最新的Loupe Browser的版本是8.1.2
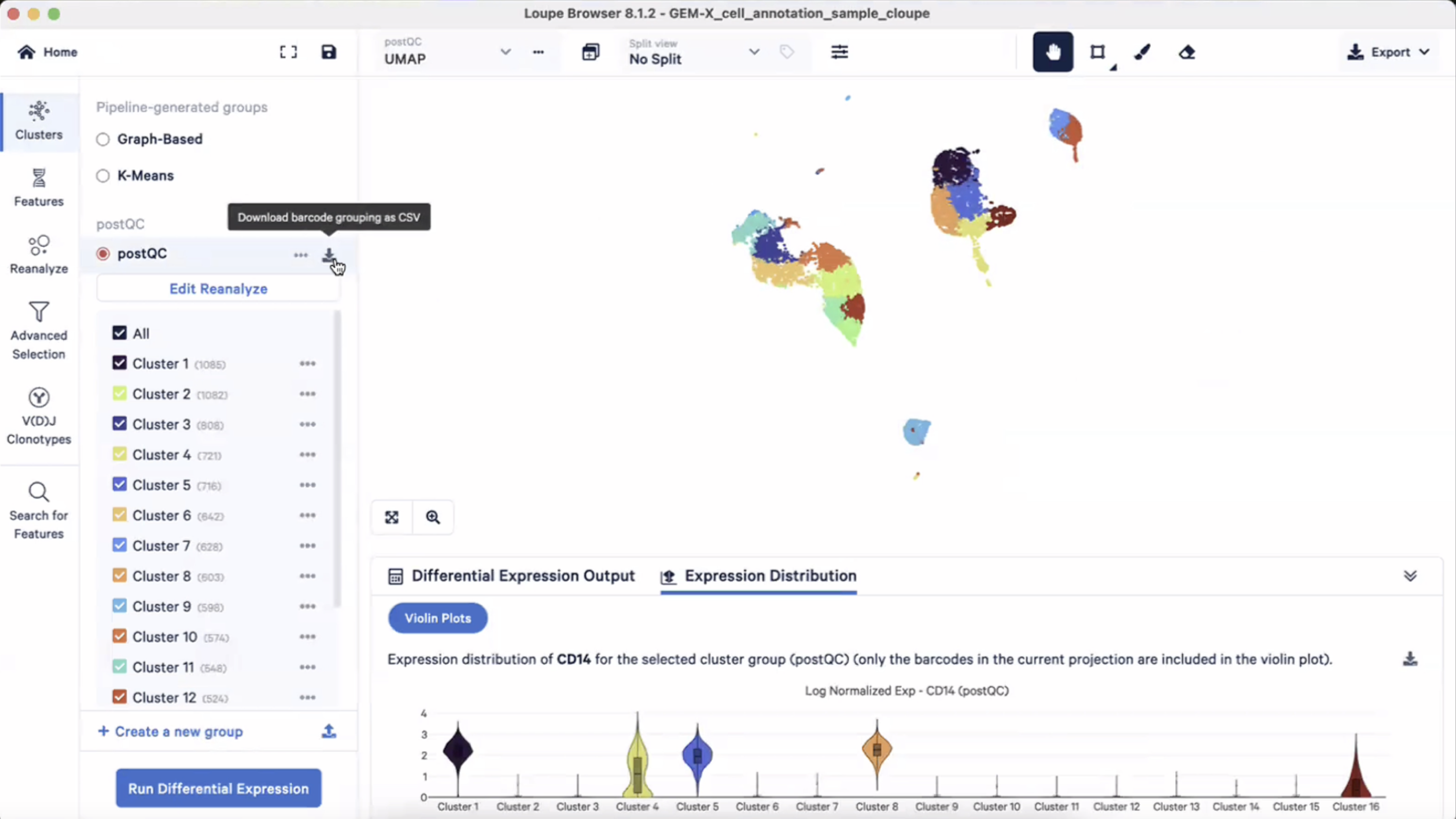
- 在用Loupe Browser打开单细胞数据后,首先看到的会是一个二维投射的结果,这个图中的每一个点都是一个细胞,不同聚类的细胞会被标记成不同的颜色,目前展示的是Graph-Based算法输出的聚类的结果
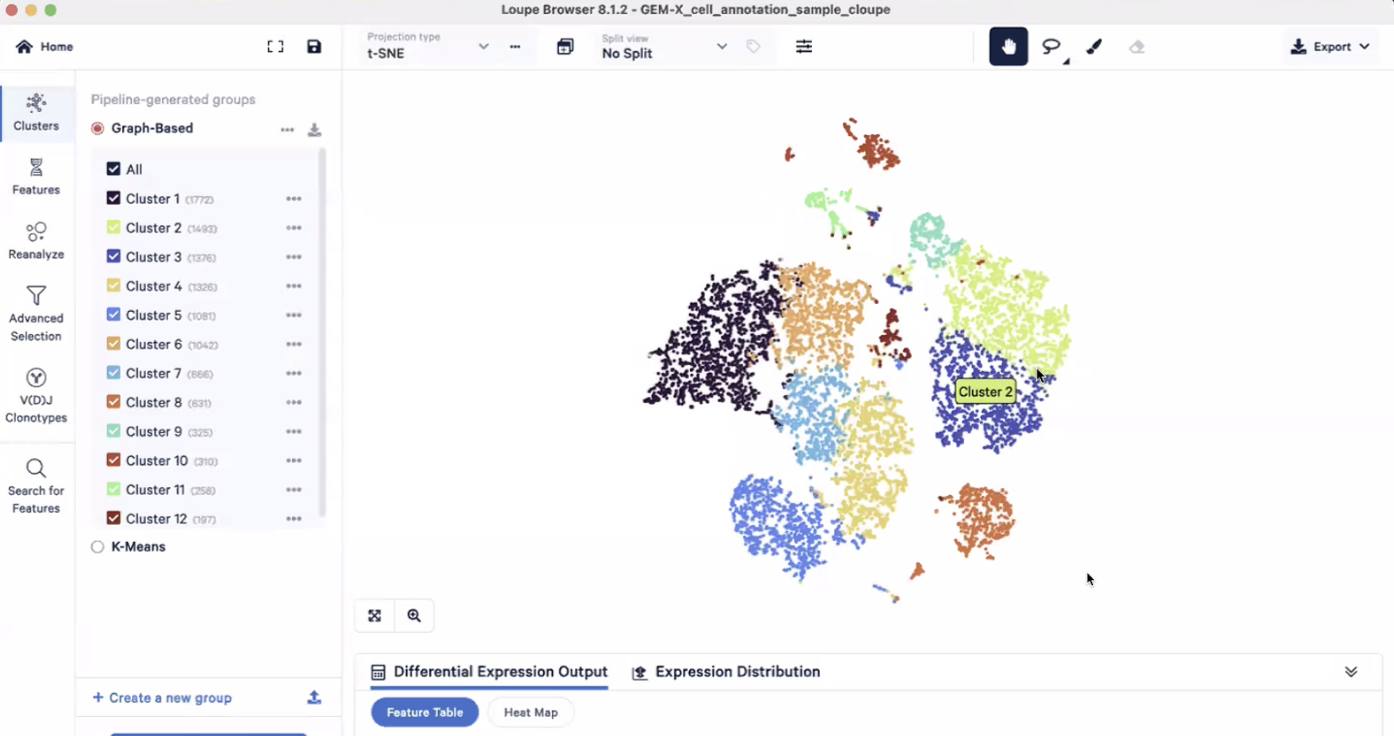
- 其他的聚类方法:比如K-Means,K值可以从2-10不等进行筛选
对页面的更多介绍
- 可以在左上角的”projection type”中选择二维投射的方法,比如tSNE或UMAP
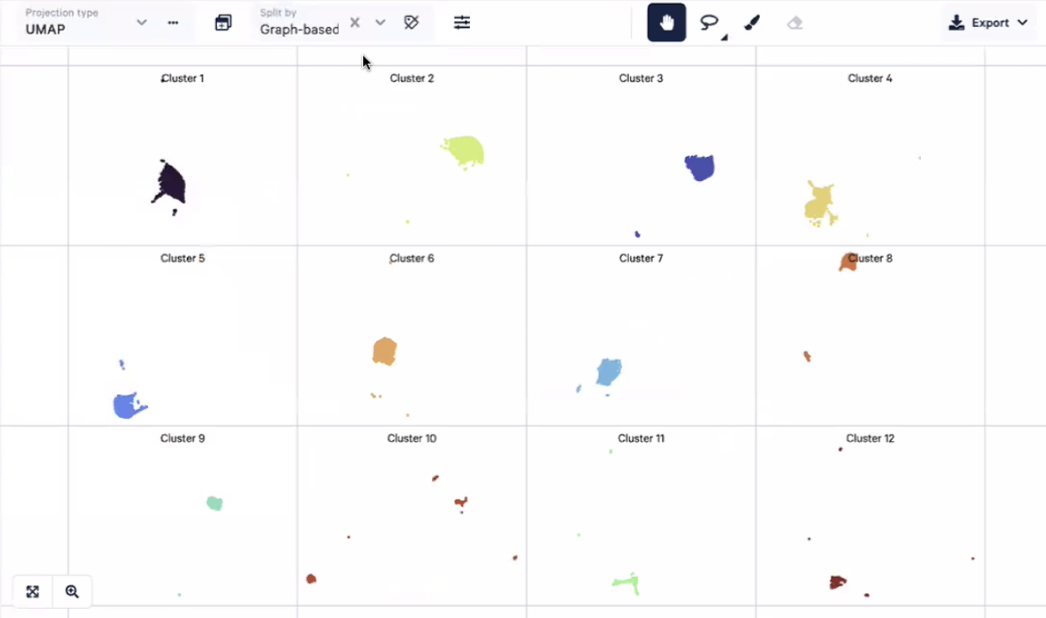
- 可以在正上方的”Split by”窗口选择细胞聚类展示的方法,比如”Graph-based”可以分开展示所有的亚群
- 右上角有各种工具可以选择细胞,比如拉索/矩形/画笔工具可以去选择我们感兴趣的细胞群,然后把这些我们感兴趣的细胞分到一个新命名的组里面去
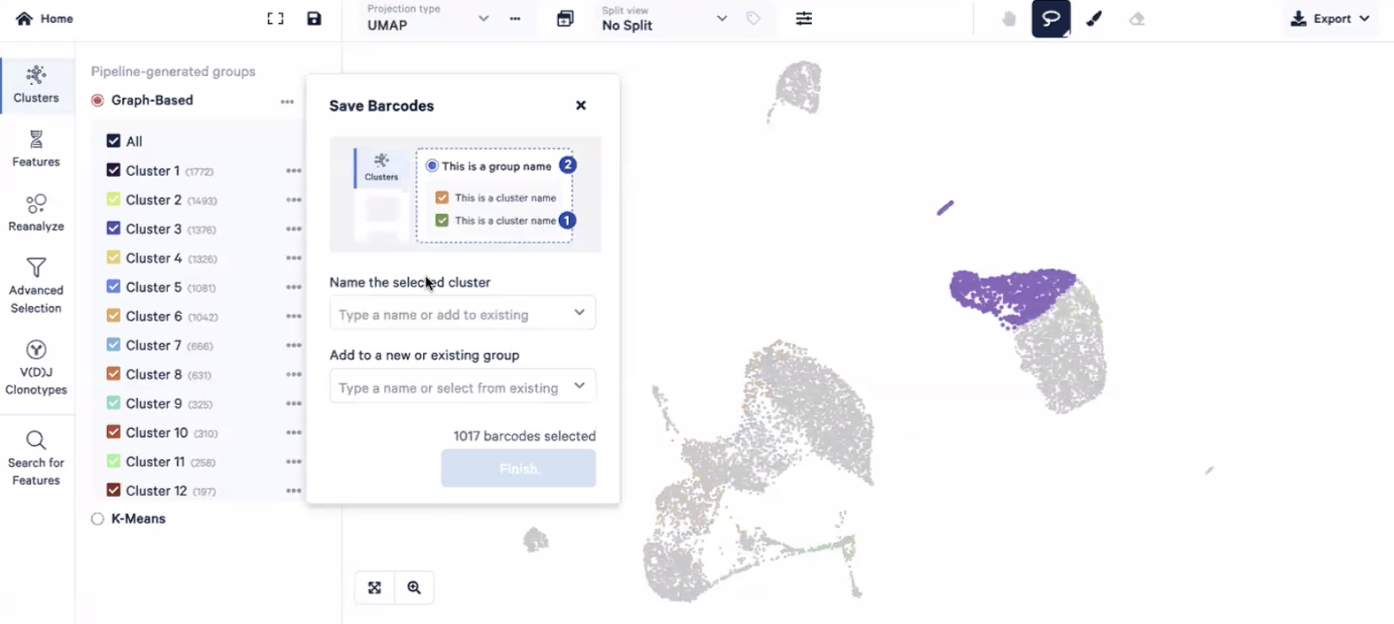
- 如果我们对当前的tSNE/UMAP图感兴趣,可以点击右上角的UMAP,把它导出成PNG或SVG的格式
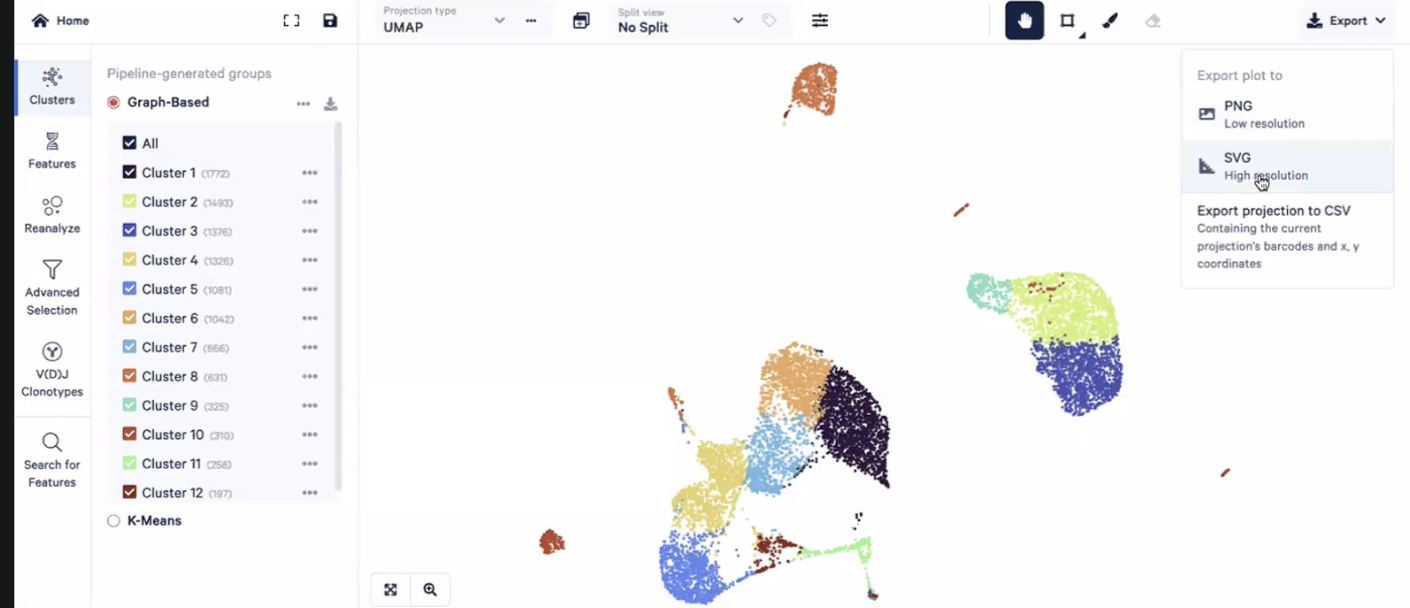
- 我们也可以把这张tSNE/UMAP图上每个点(对应一个细胞)的坐标保存成CSV格式的文件
app左侧的功能栏
- 图片的左侧有不同的功能栏,其中cluster可以给我们展示细胞分群的结果,我们打开软件后看到的cluster是Cell Ranger自动生成的,也被自动地放入了cloupe文件中,我们之后也可以不断添加别的信息在这里面
- 第二个功能是features,在这里我们可以去展示我们感兴趣的基因在整个数据里面的表达情况
- 比如我们对CD3E这个基因比较感兴趣,那我们就会发现每个点被渲染上了不同的颜色,从浅红色到深红色,其中颜色越深说明细胞的表达量越高
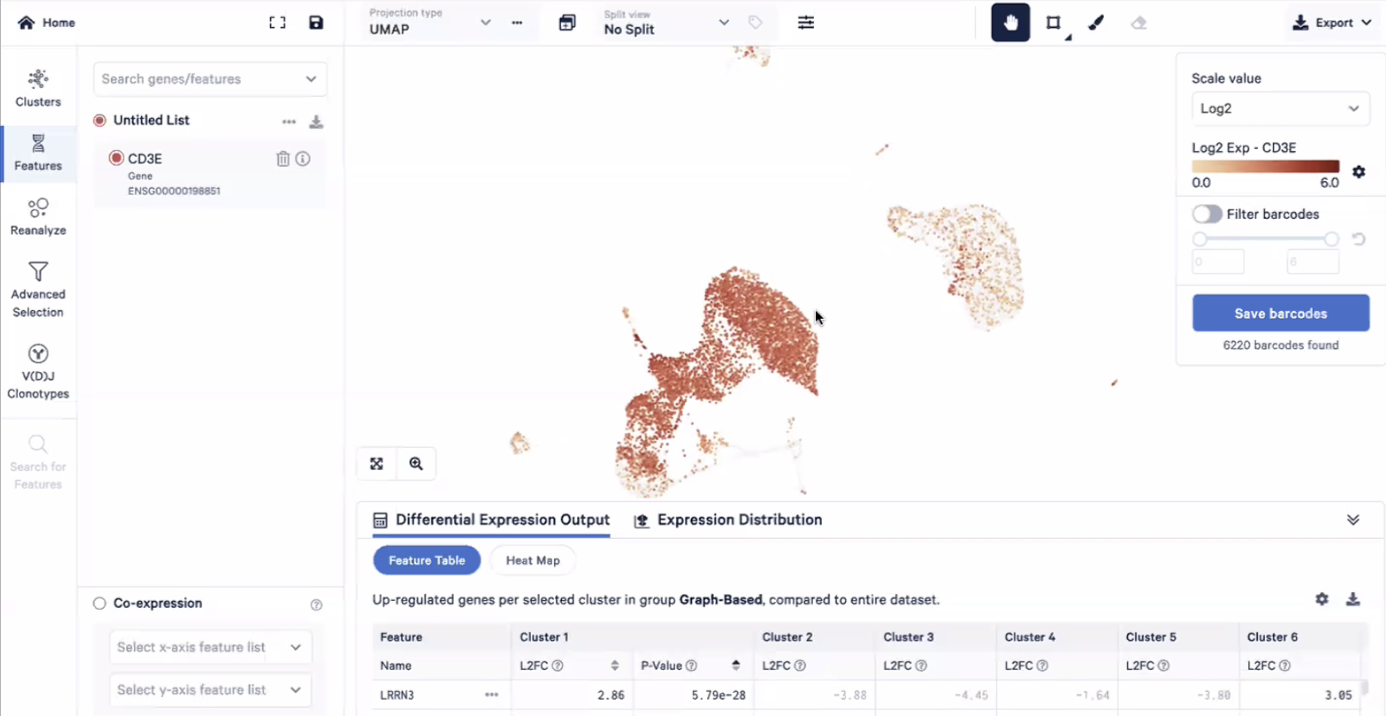
- 上图中展示的是每个细胞内细胞的捕获数目做Log2的转换,如果想更加直观地看到每个细胞被捕获到的转录本的具体数值,可以选择Linear
- 如果想要比较不同细胞之间的,我们通常需要把整个捕获到的转录本进行均一化/标准化,也就是Normalization,而LogNorm是一种标准化的形式,我们可以通过转化成LogNorm去直观地比较某个基因在不同细胞之间的表达差异
- Reanalyze可以用于简单的质量控制QC,见下
- Advanced Selection:通过创建规则,去更准确地选择我们感兴趣的细胞
- V(D)J的数据:对于要关注免疫细胞的老师可以用到
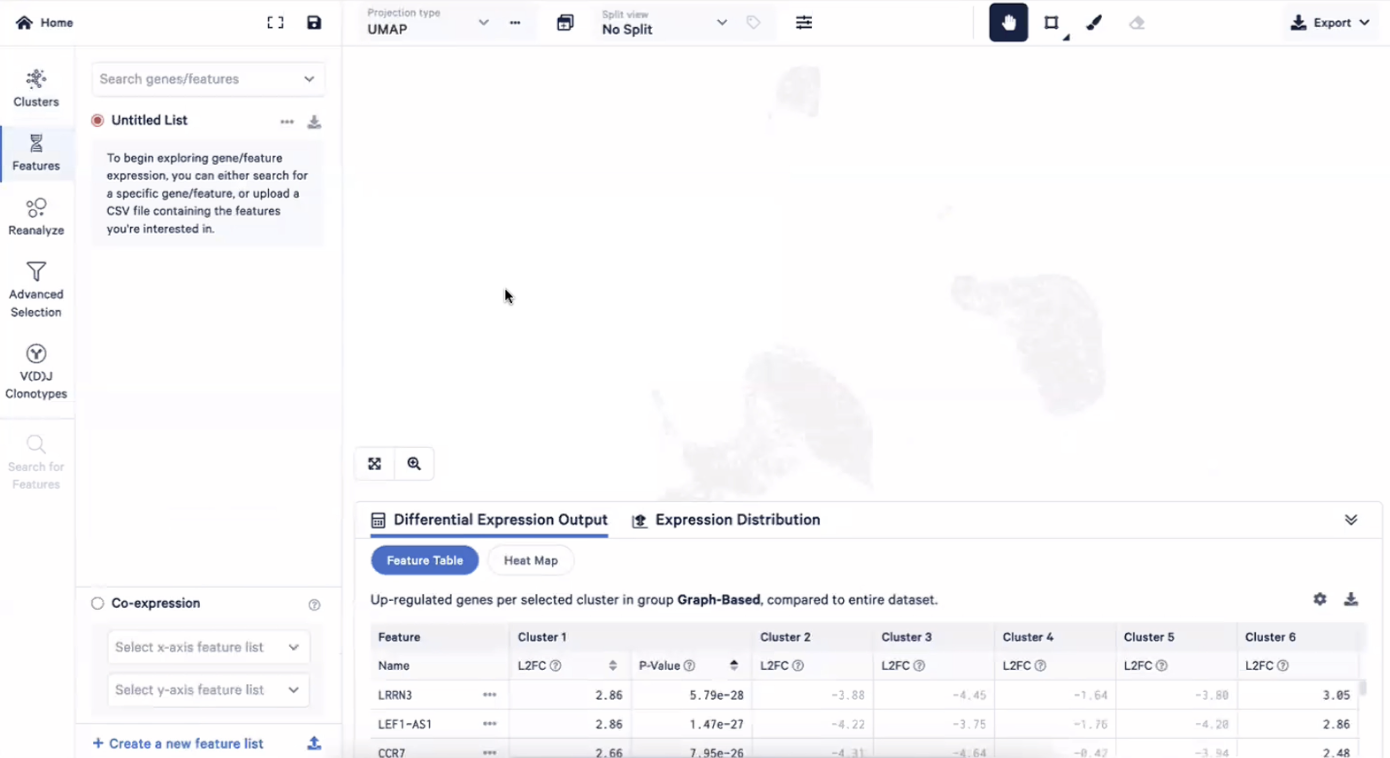
- 各种各样的表格,首先我们看到的是差异表达基因的表格,这里展示的是每个cluster的marker基因,是把所有cluster1的细胞和所有非cluster1的细胞进行比较的,然后计算出Cluster1里面到底富集了哪些细胞,可以对Log2FC和矫正后的P值进行排序。
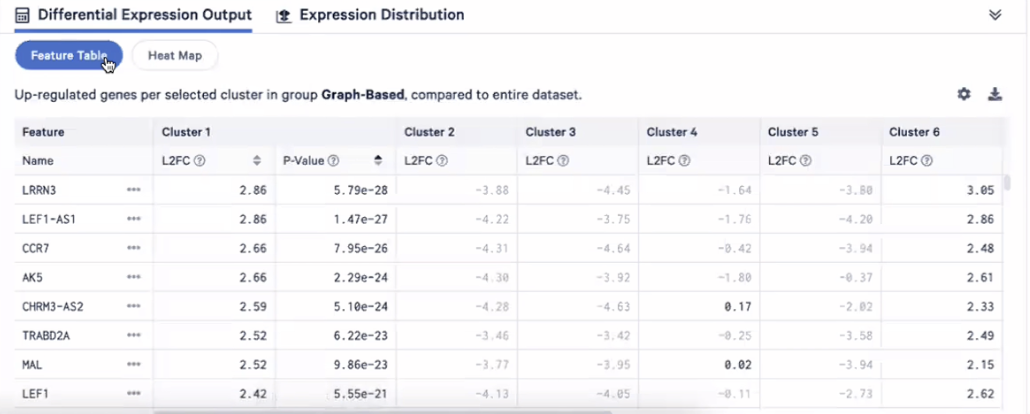
- 我们可以用热图的方式展示上面的表格,每一行是一个cluster,每一纵列是一个基因,颜色代表的是基因的表达差异倍数变化
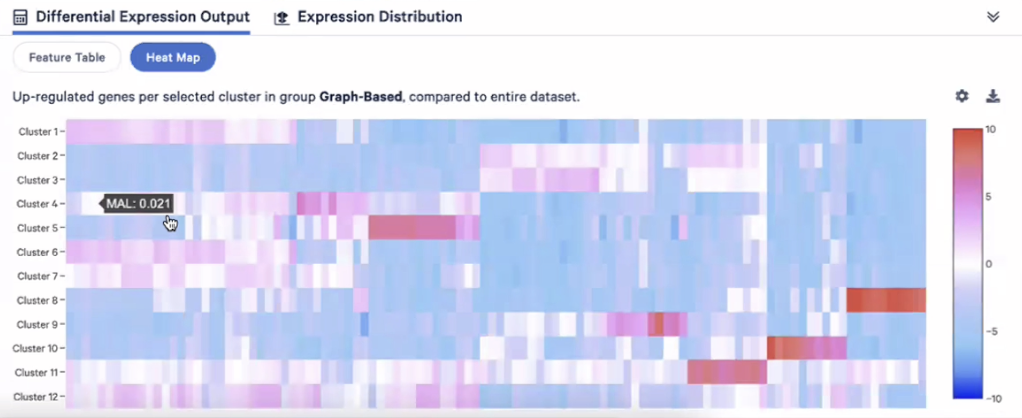
- 我们也可以查看某一个基因在每个聚类的基因表达情况,这里展示的是CD3E基因,表达差异情况是LogNorm格式展示的,这里我们可以发现CD3E在Cluster1、4、6、7高表达,那我们就可以说Cluster1、4、6、7很有可能是T细胞
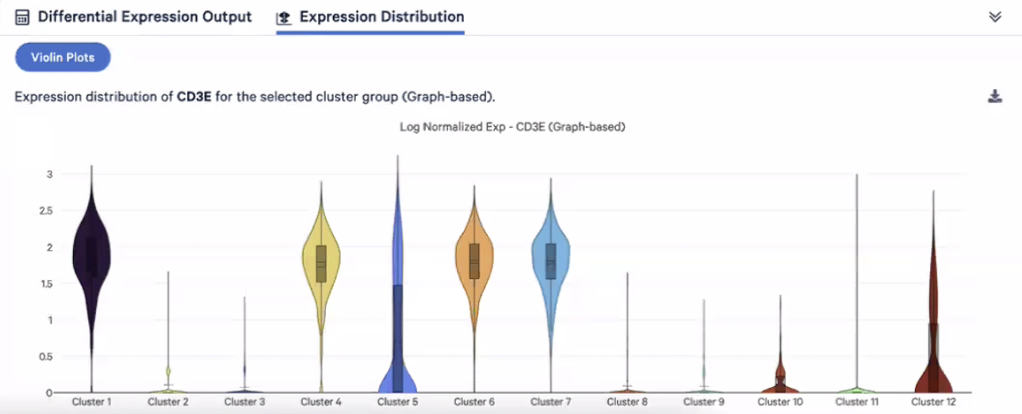
- 前面提到,我们可以通过QC去过滤掉低质量的细胞,我们点击Reanalyze来进入这个部分
- 在点击了Reanalyze之后,我们可以看到新的窗口弹开,我们会在这个新的页面中进行QC
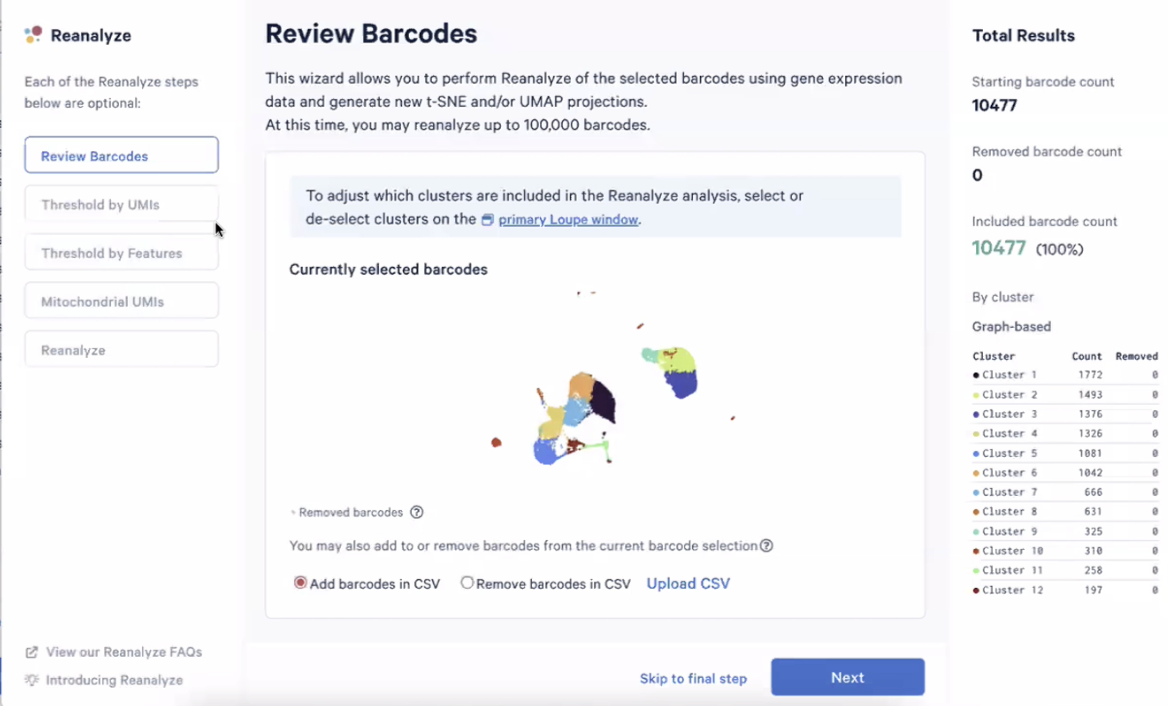
- 在QC中,我们主要基于三个指标,分别是每个细胞内探查到的UMI数目(转录本数),每个细胞内的基因总数,以及每个细胞内的线粒体基因百分比,目前我们有10477个细胞被选中,在QC的过程中,细胞的数量会不断减少
- 首先,我们会对细胞中的UMI总数进行QC,在这里请避免套用其他人的分析指标进行分析,比如Seurat的教程QC指标
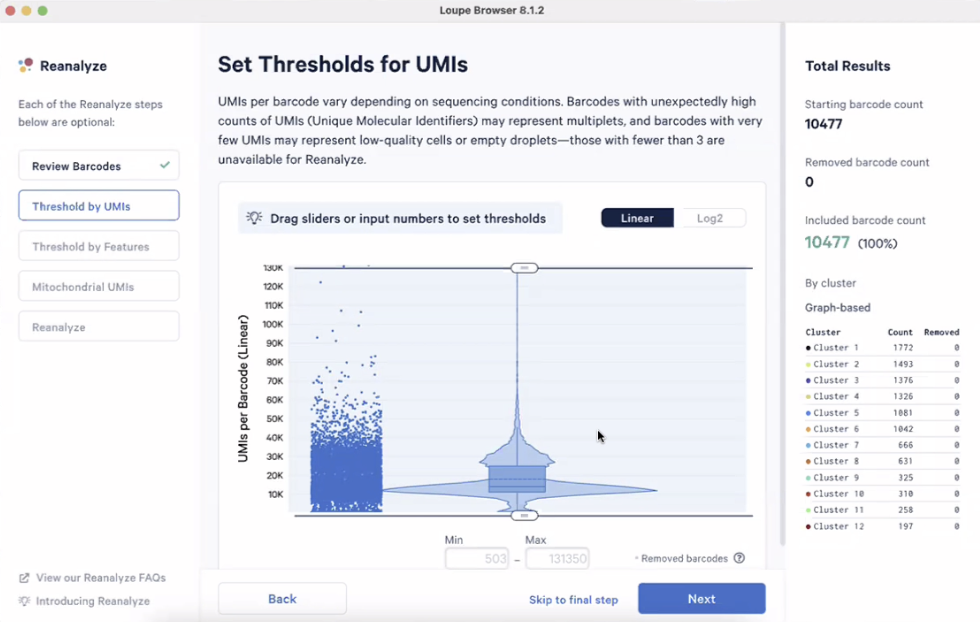
- QC的目的是去除那些离群的细胞,我们希望找到一个合适的区间,能让大部份的细胞都落在这个区间之中,不同的数据和样本中,这个区间的位置也会出现变化的,因此不可以使用别人的区间去套用在自己选择的数据上
- 通常来说我们会建议大家观察整体的数据分布情况,比如每个细胞内的UMI数量是怎么样的,对于我们的数据去取上限和下限值
- 可以看到Y轴是每个细胞内探测到的转录本总数,旁边是小提琴图取展示分布,我们可以对纵轴的数据进行Log2转换
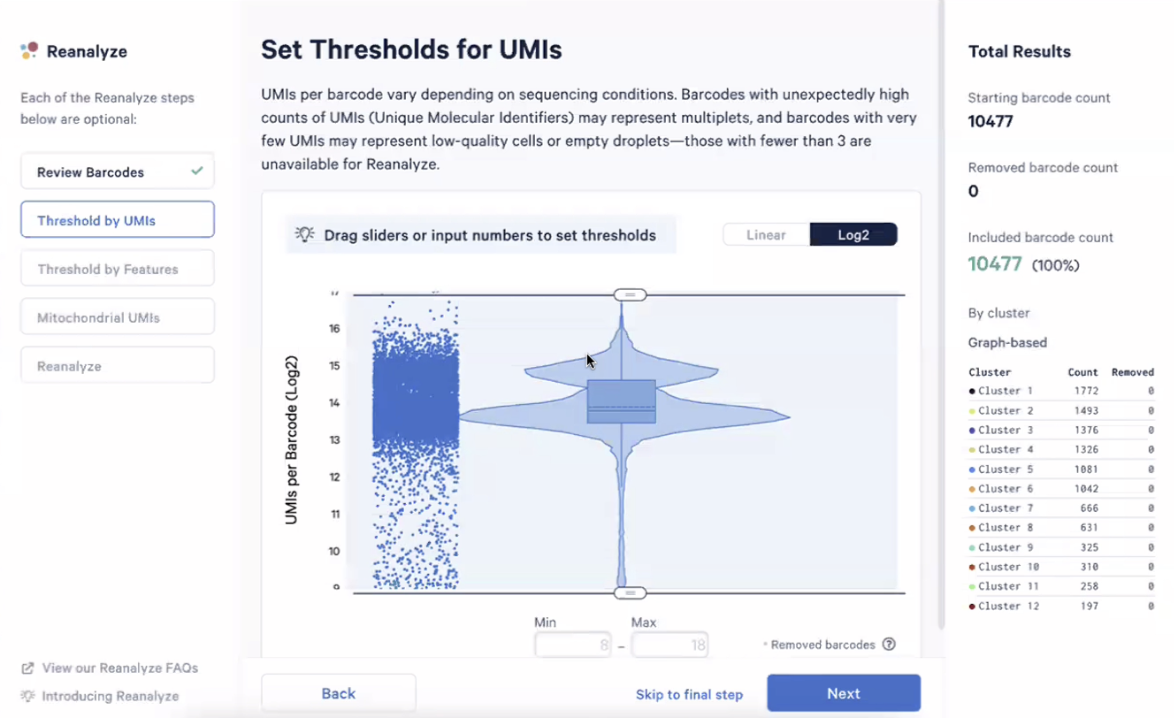
- 这里是最新的GEM-X设备上产生的PBMC数据,之前版本的数据(比如3’-v3,5’-v2)通常不会看到双峰结构,不用担忧,这并不是展示的GEM-X的数据出现了问题,而是说明GEM-X的敏感性非常高,高到能把两种细胞亚群(髓系和淋系)明显地分开来。
- 回到QC,我们希望把离群的,低于某个下限值的细胞去掉(这些细胞可能已经死了或快死了),同时也把多余某个上限值的细胞去掉(这种情况下可能一个液滴里有两个细胞),我们也可以手动输入某个值。
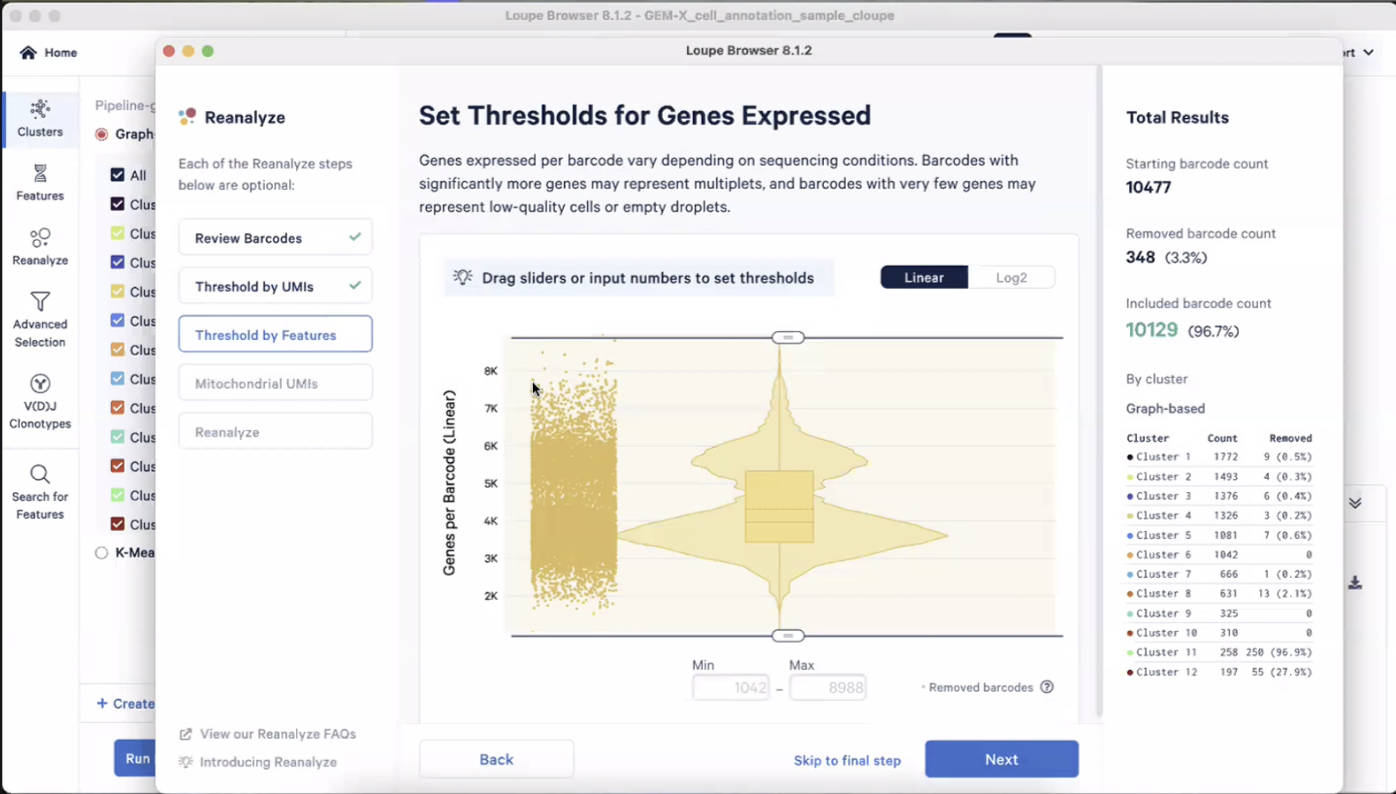
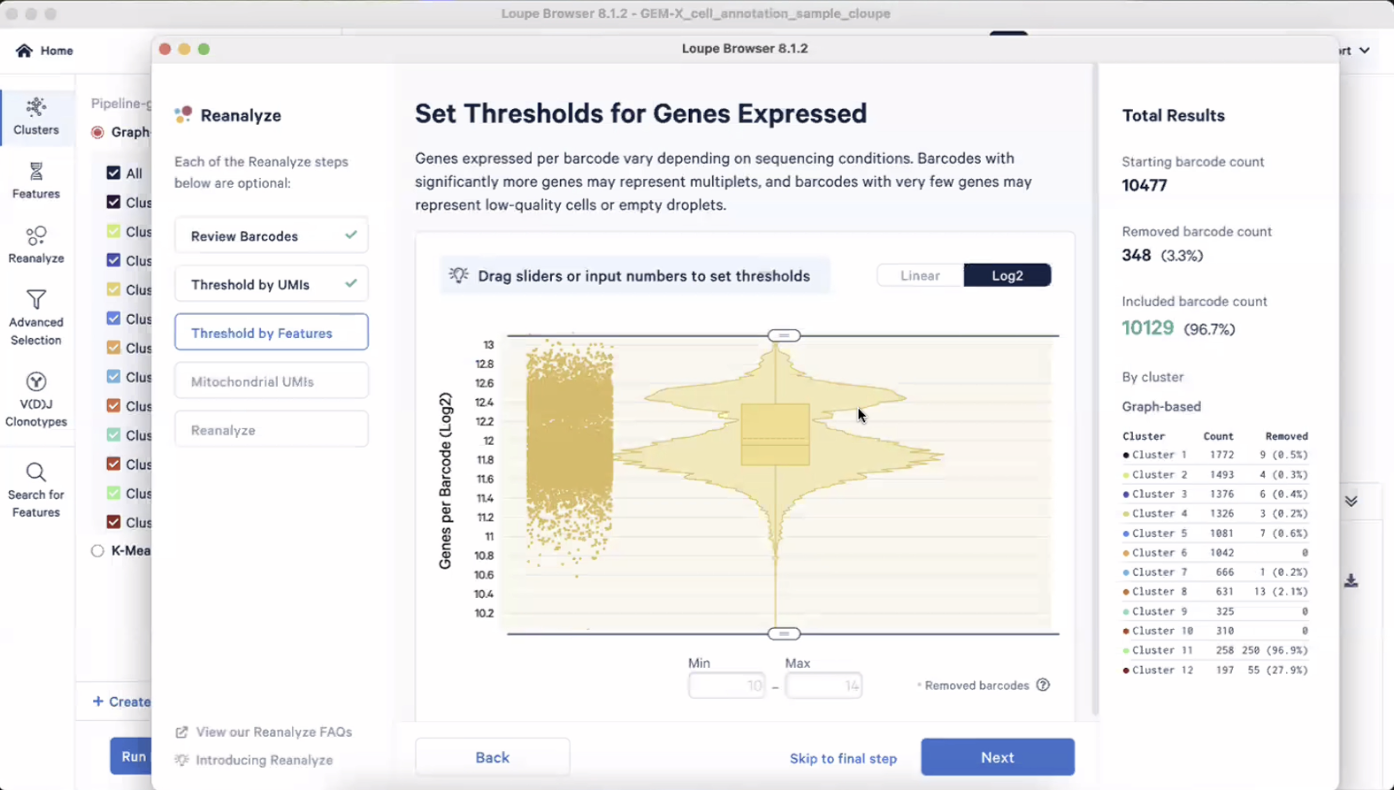
- 接下来,我们针对每个细胞内捕获到的,总的基因数进行QC,我们也可以对纵轴数据(基因数)进行Log2转换,也能看到双峰结构,我们也可以通过之前说到的方法进行QC
- 最后,我们可以针对每个细胞内捕获到的线粒体基因的百分比进行QC,如果一个细胞处于将死状态,细胞膜破裂了,细胞内的RNA就会释放出来,对于这样的细胞,其总的RNA数目就会下降,但是相对之下,线粒体内的RNA是保留在线粒体的双层膜结构中的,线粒体的RNA保留会相对更好,也就是分母的总RNA数下降,而分子的线粒体RNA数目不变,总的百分比就上升了,这就是为什么高的线粒体基因百分比可能是一个不太好的指标。具体操作如下:
- 首先我们要告诉Loupe Browser我们要研究的这群细胞的染色体基因名称,如果是研究人或者小鼠,Loupe Browser中就已经有内置的小鼠线粒体基因信息了,如果研究其他物种,可能需要准备自己的线粒体基因的名字
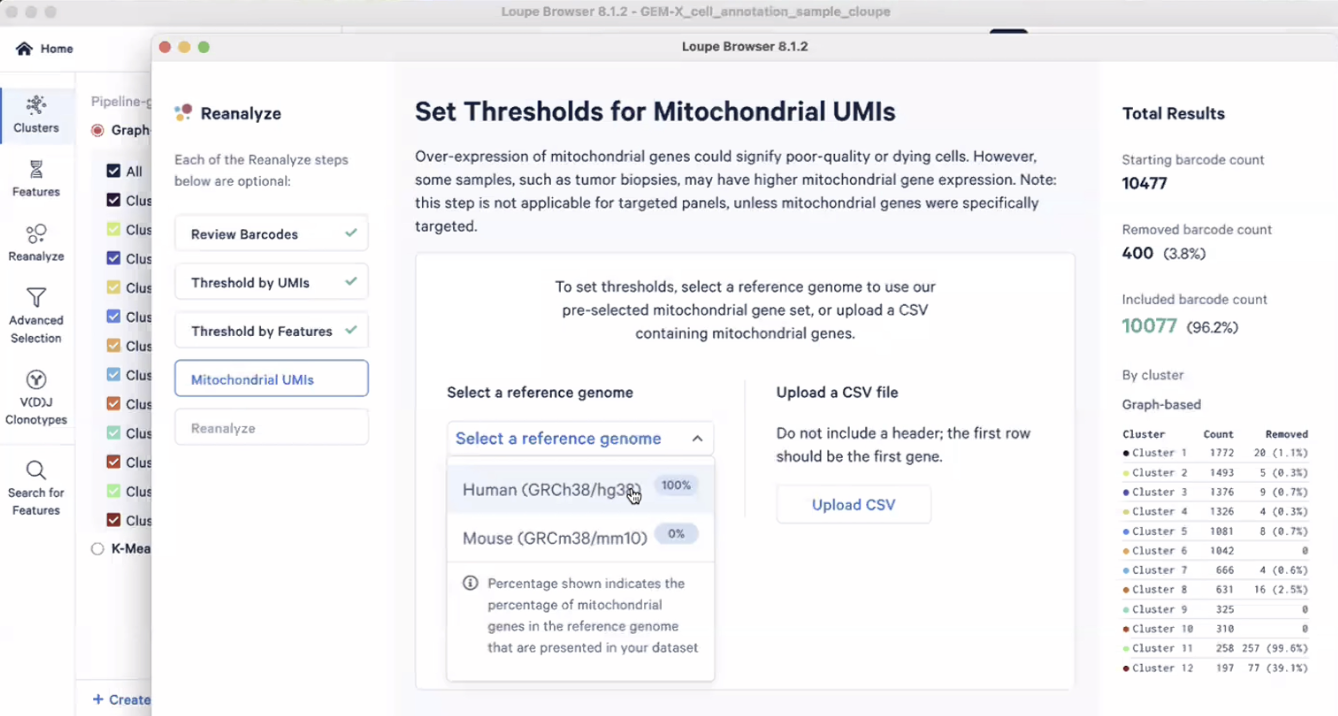
- 我们可以得到一样的图,但是图里面变成了线粒体基因的百分比,这一> 来源:步我们会想去除那些线粒体基因占比过高的细胞,因为这些细胞是将死的细胞
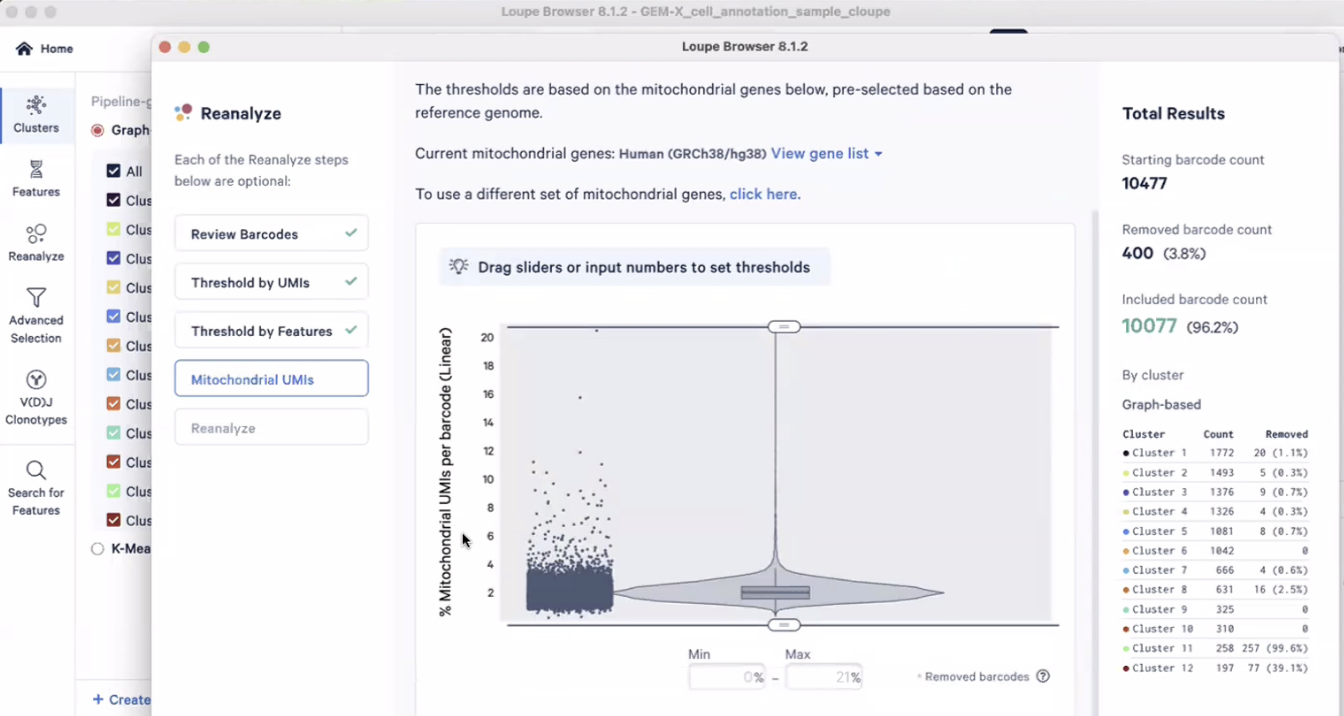
- 在确定了线粒体基因的百分比上限为5%(手动确定,仅作示例,不构成参考,详见备注)之后,我们剩余的细胞还有10004个
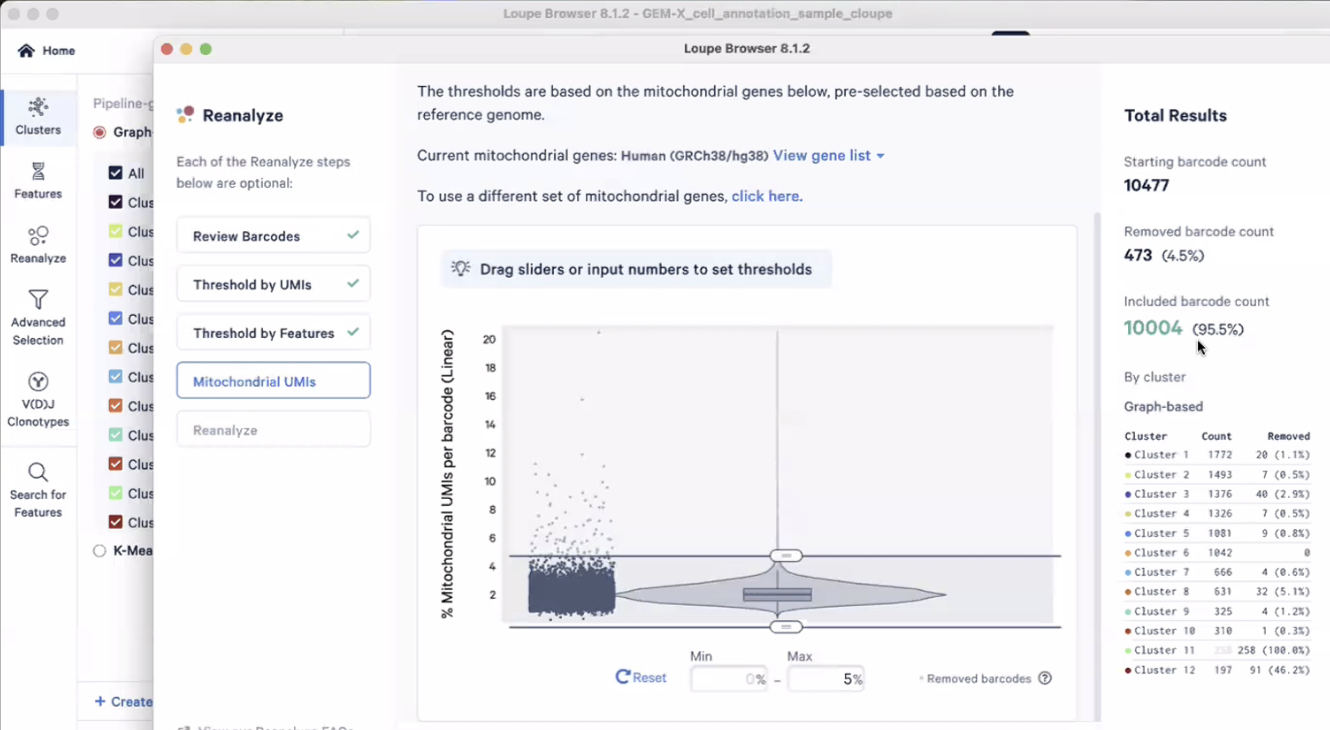
- 我们对这些细胞重新计算tSNE和UMAP,重新计算cluster,我们把这10004个细胞重新命名为一个样本,比如postQC,在这个运行的过程中请不要关闭窗口
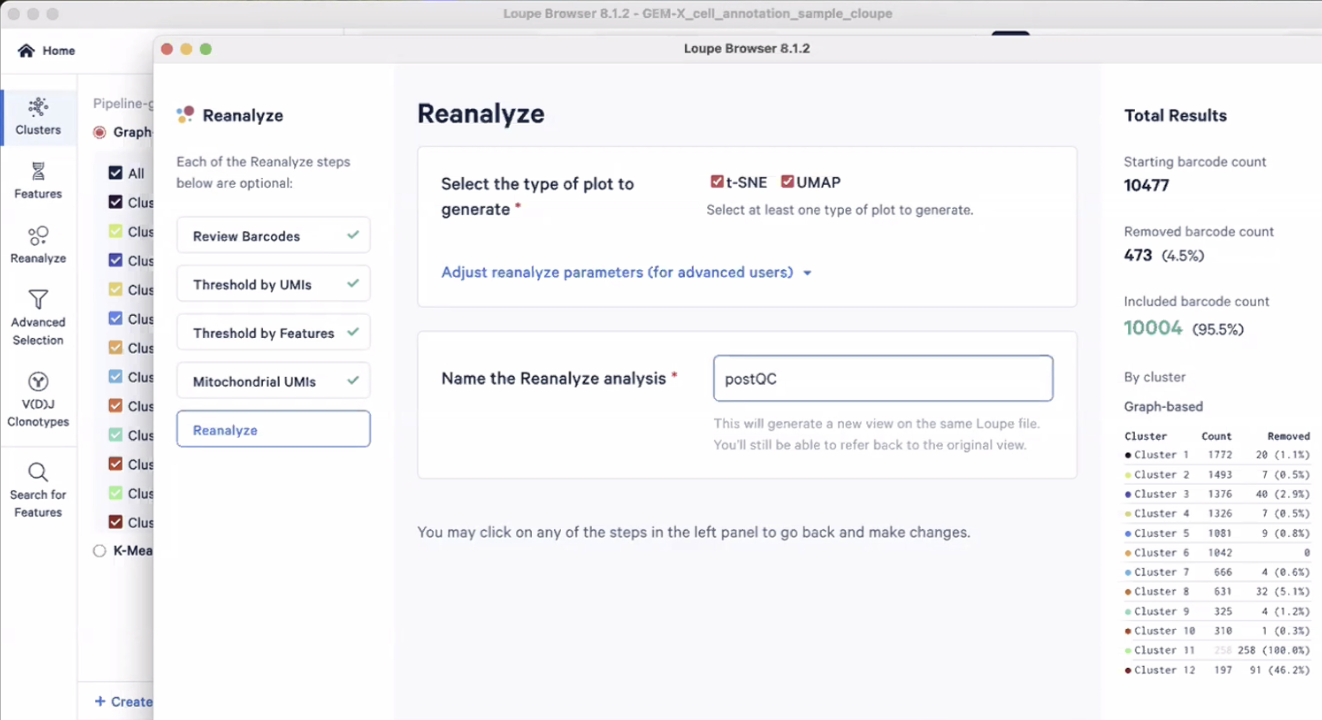
- 在完成了QC后,我们可以发现在cluster页面出现了新的组别postQC
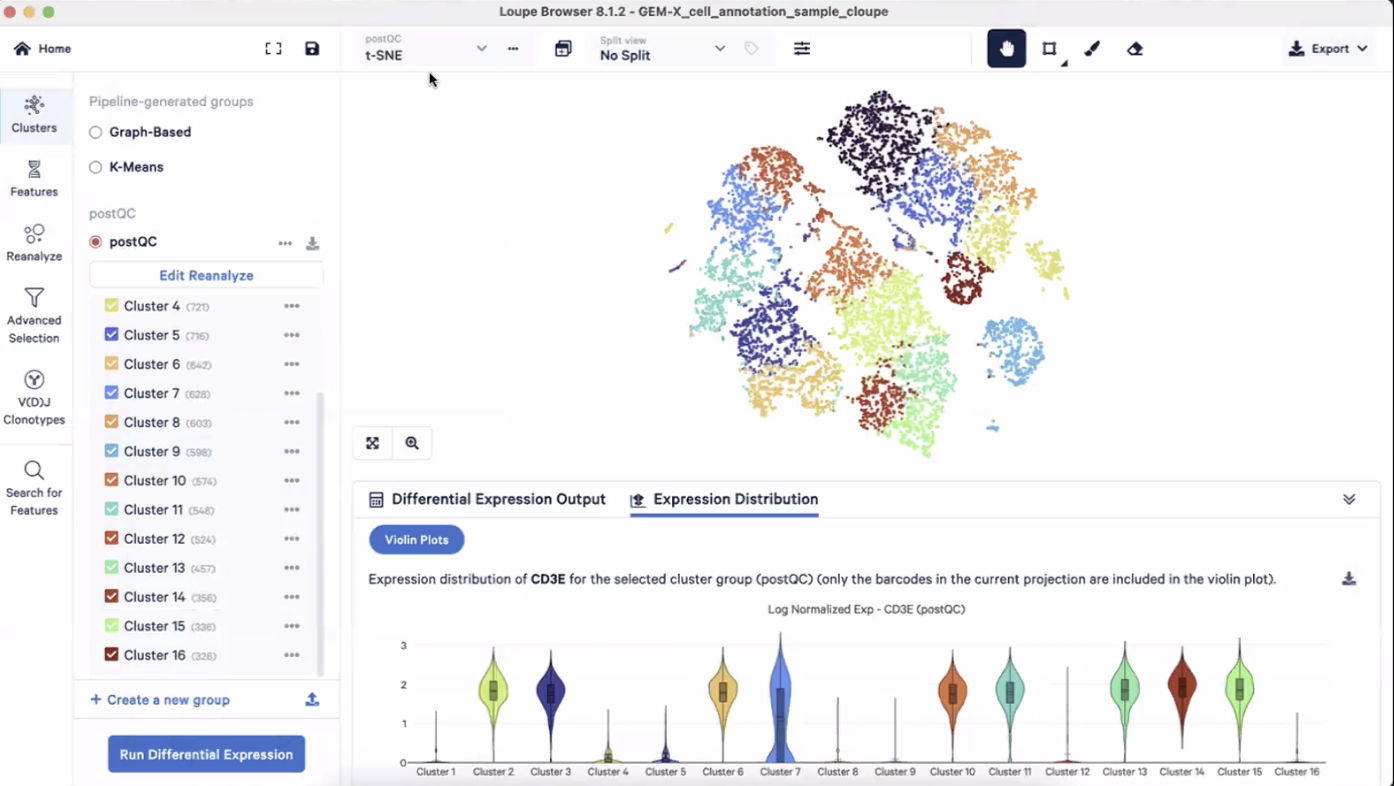
- 投射结果也可以选择postQC的tSNE或UMAP
细胞注释
细胞注释的几种方式
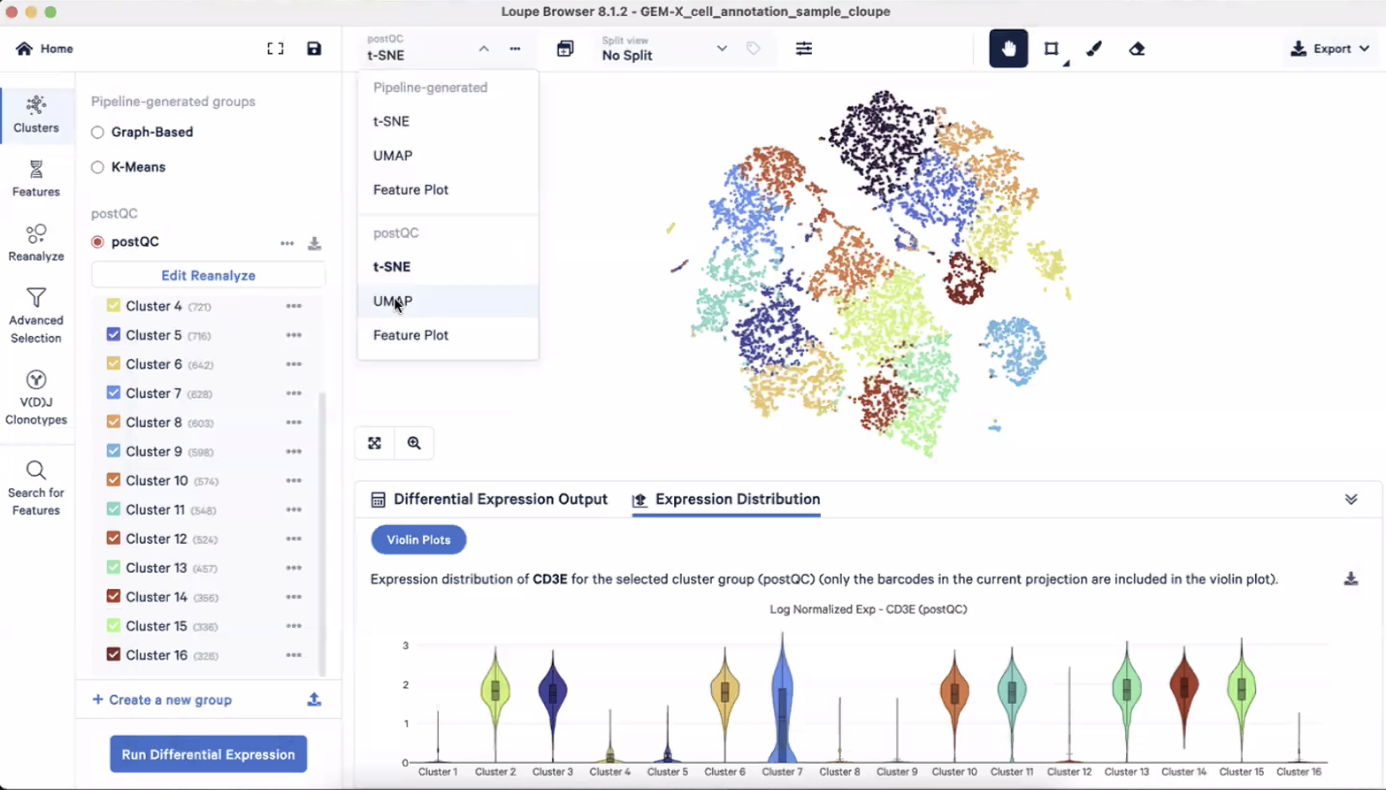
- 去计算每个cluster的marker基因,在这个过程中,我们会对每个cluster进行差异表达基因分析,找到每个cluster的marker gene是什么,点击”Run Differential Expression”,组别选择第二个”Between selected cluster(s) themselves”,然后就可以开始计算每个cluster里富集的marker gene是什么,通过观察每个cluster里富集的marker gene,我们就可以注释每个cluster的细胞类型,这种注释方法是所谓的“unbiased and unsupervised”(文献中常见)
- 第二种细胞注释:比如我们对自己的样本非常了解,样本中有哪些细胞类型早已烂熟于心,细胞类型的marker gene也很熟悉了,那我们就可以直接把marker gene罗列出来,看下每个marker gene在每个cluster里是怎么样的,比如在这个PBMC的case中,如果我们希望观察髓系的cluster,那我们可以输入CD14这样的marker gene,然后看看CD14在哪一些cluster中高表达
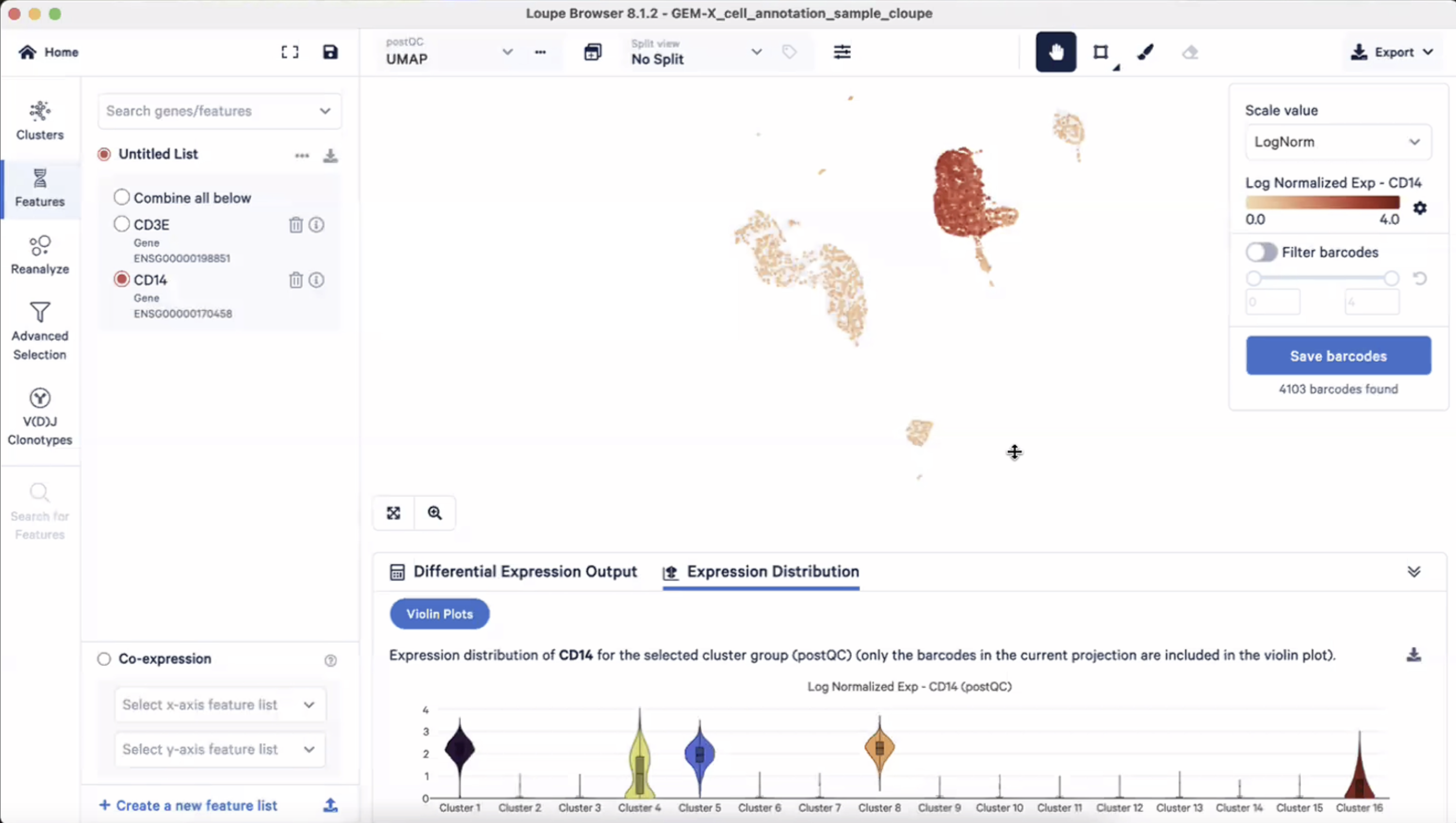
- 在上图中,我们可以看到CD14在Cluster1、4、5、8、16中高表达,因此我们可以猜测这五个cluster可能是髓系来源的,我们这时可以在advanced selection里把这五个cluster进行选中
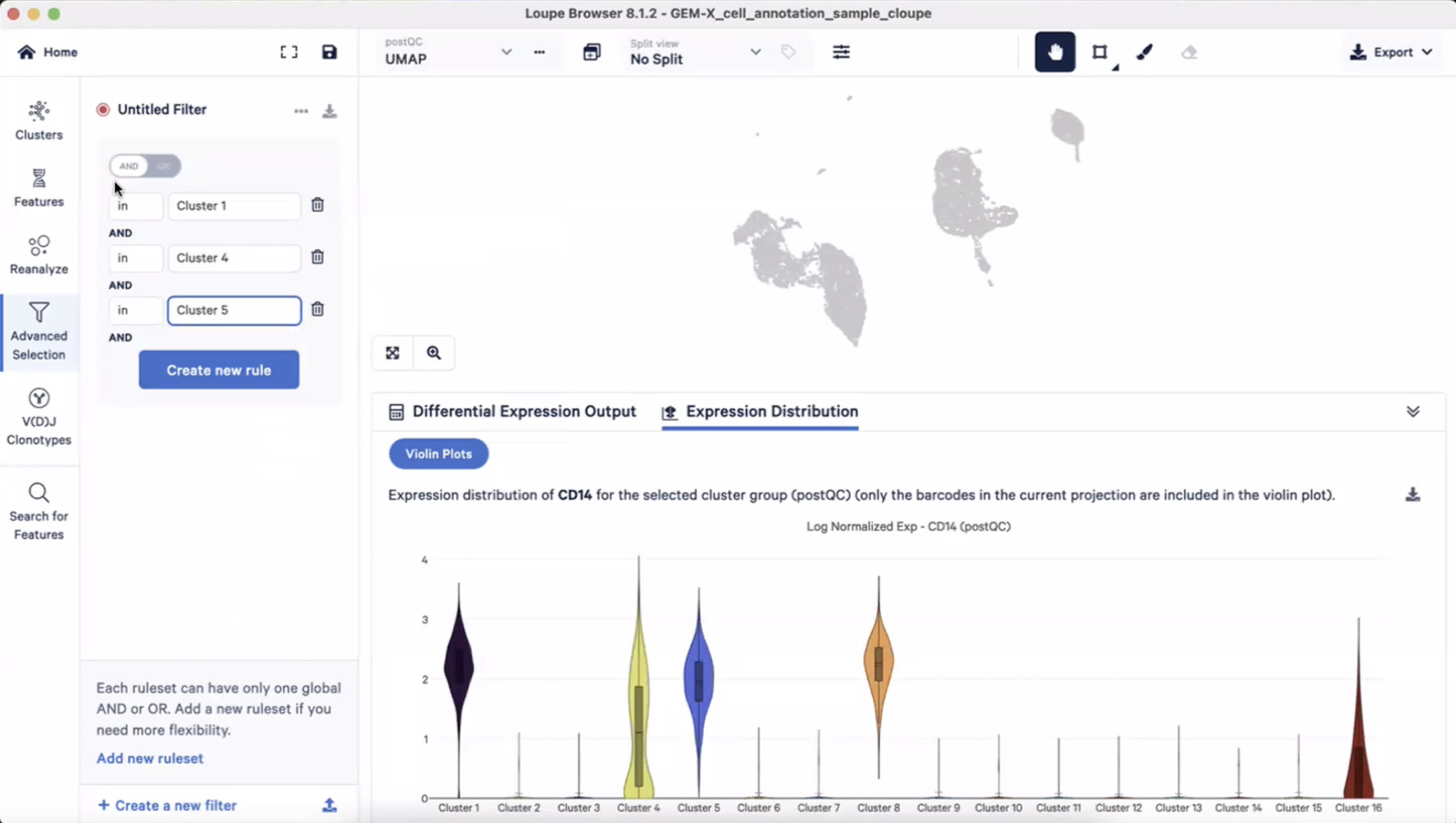
- 我们在选择的过程中,要注意鼠标上方的“逻辑开关”,把判定条件设为”OR”,这样我们就选出了自己感兴趣的5个cluster
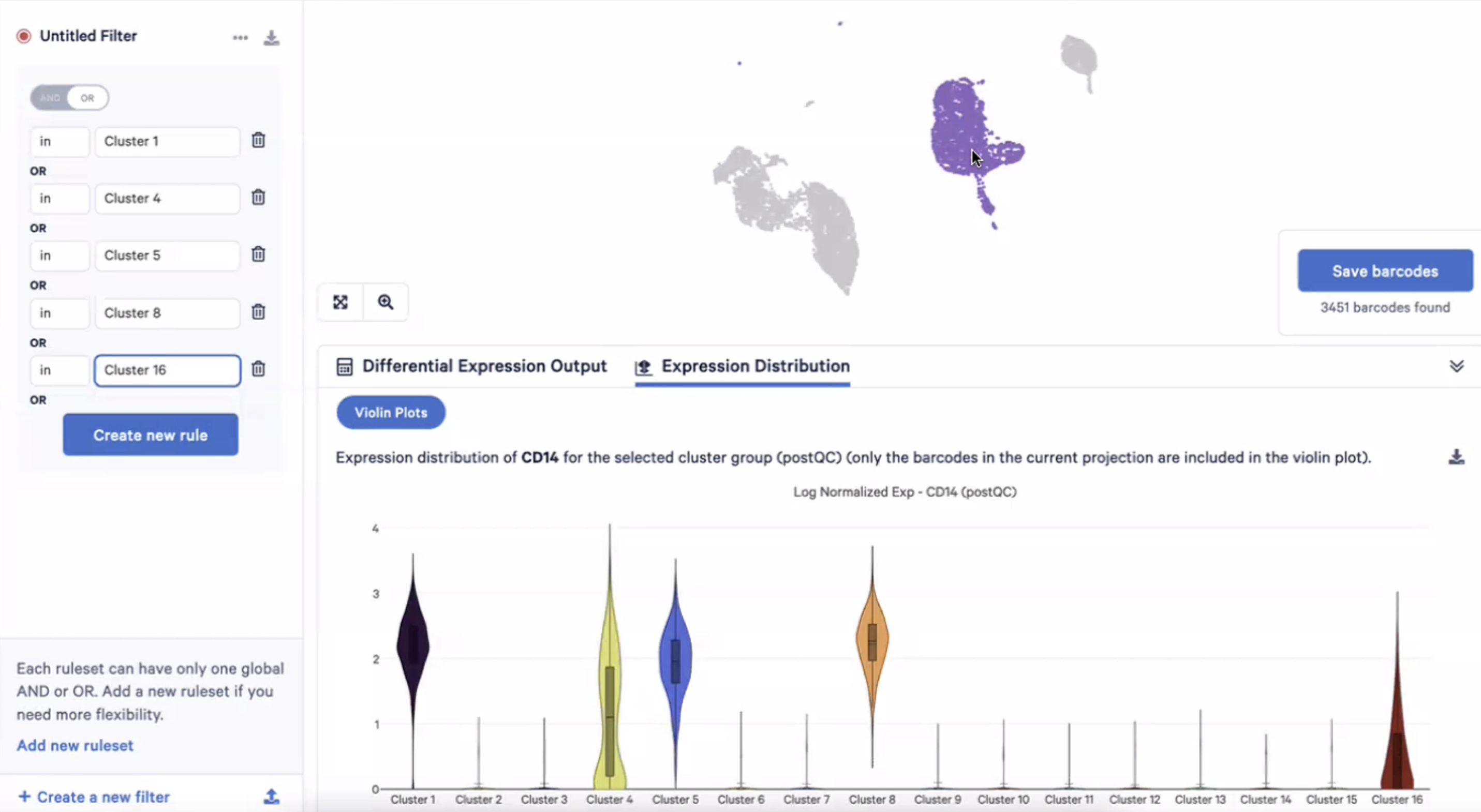
- 我们可以把这些细胞进行保存,命名为一群新的细胞“髓系myeloid“,并且把数据分组归到新的组别”细胞注释cell annotation“中
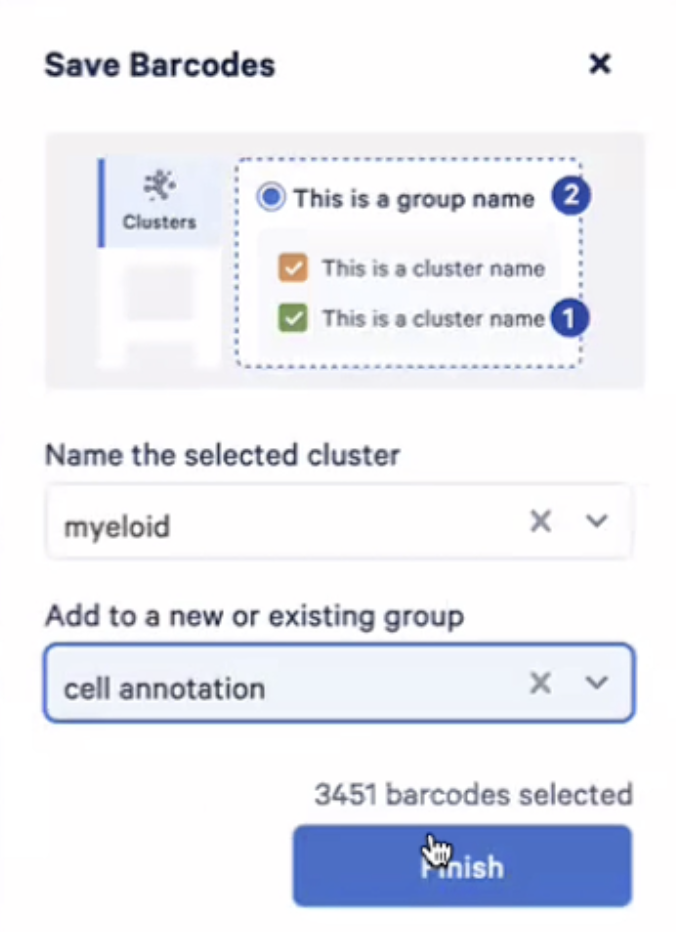
- 我们可以通过这种方式,人工地通过对每个cluster已知的marker gene进行注释
- 第三种方法是细胞自动注释,比如说我们研究的样本恰好是与前人研究过的某些类型相类似的,发表了文章,也有对应的单细胞数据和注释结果,我们可以“借用”他们的单细胞数据,然后自动地把我们的单细胞数据进行注释
- 目前来说自动细胞注释在Loupe Browser中仍无法实现,需要用到一些三方软件,比如Seurat,这个工具中有label transfer的算法,通过铆定点的方式去transfer,实现自动的细胞注释
数据保存
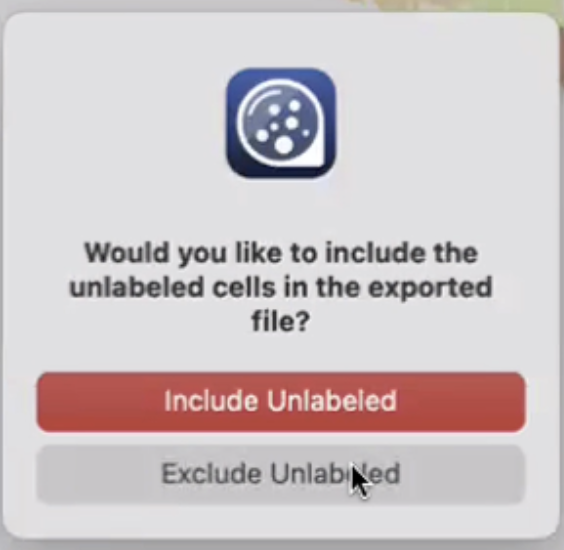
- 如果我们对QC后的数据比较满意,我们可以点击postQC组旁边的下载按钮,将数据以csv的格式保存在本地,注意选择”exclude unlabeled”来去除低质量的细胞
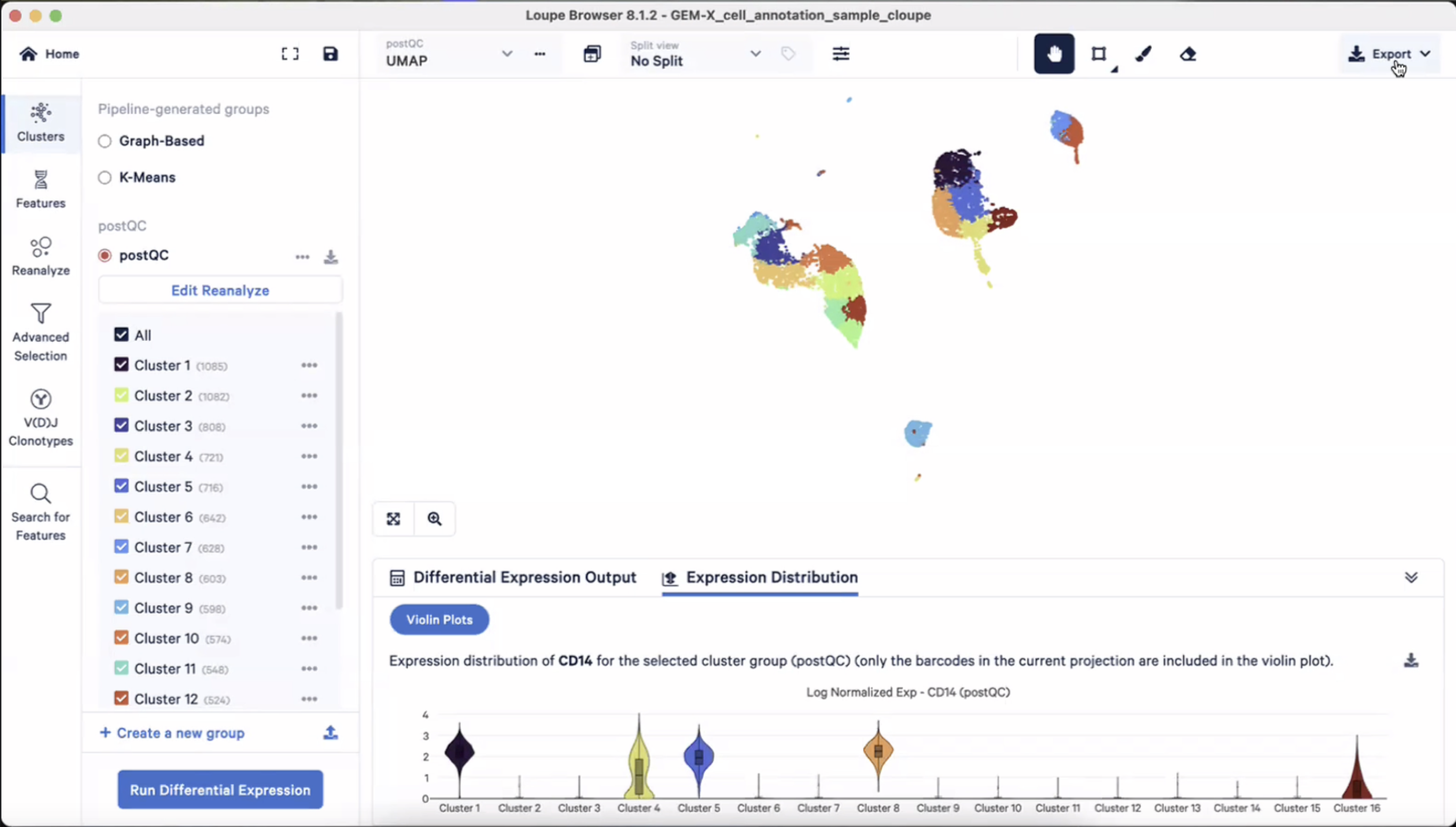
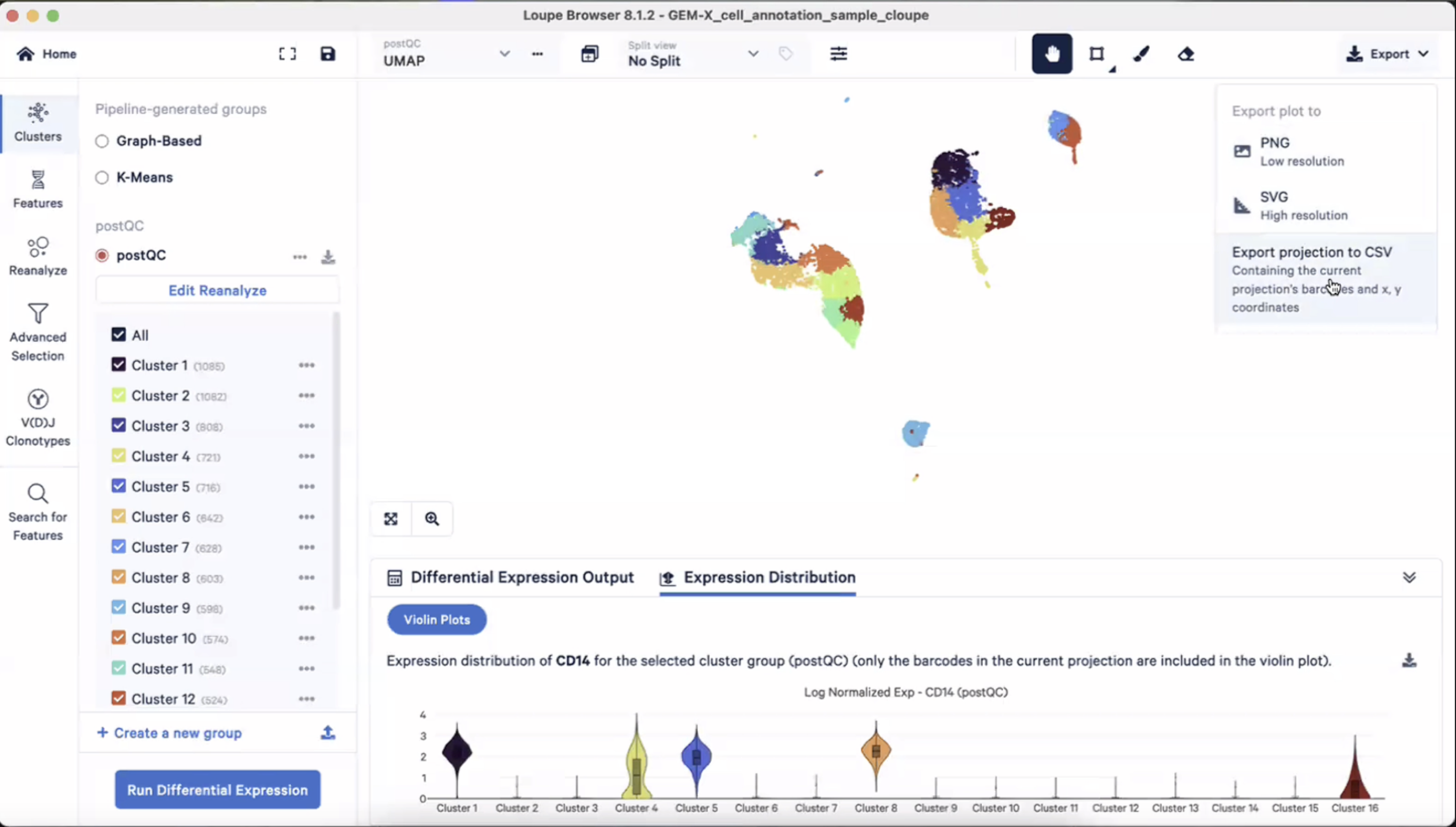
- 保留UMAP:右上角的Export按钮-“Export projection to CSV”
Loupe R
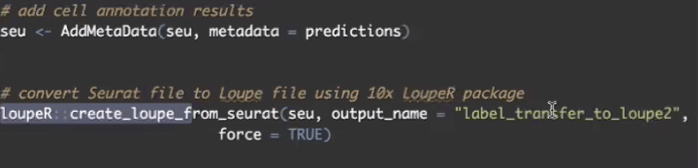
- 在经过了Seurat的自动注释后,我们可以通过Loupe R将注释后的Seurat数据转回cloupe文件,进行下一步的分析
- 对于自动注释,我们要把Cell Ranger产生的细胞基因矩阵,也就是filtered_feature_bc_matrix.h5文件,这个文件可以自动被Seurat读取
- 把UMAP数据导入Seurat(下面还有把postQC的clustering结果导入Seurat)
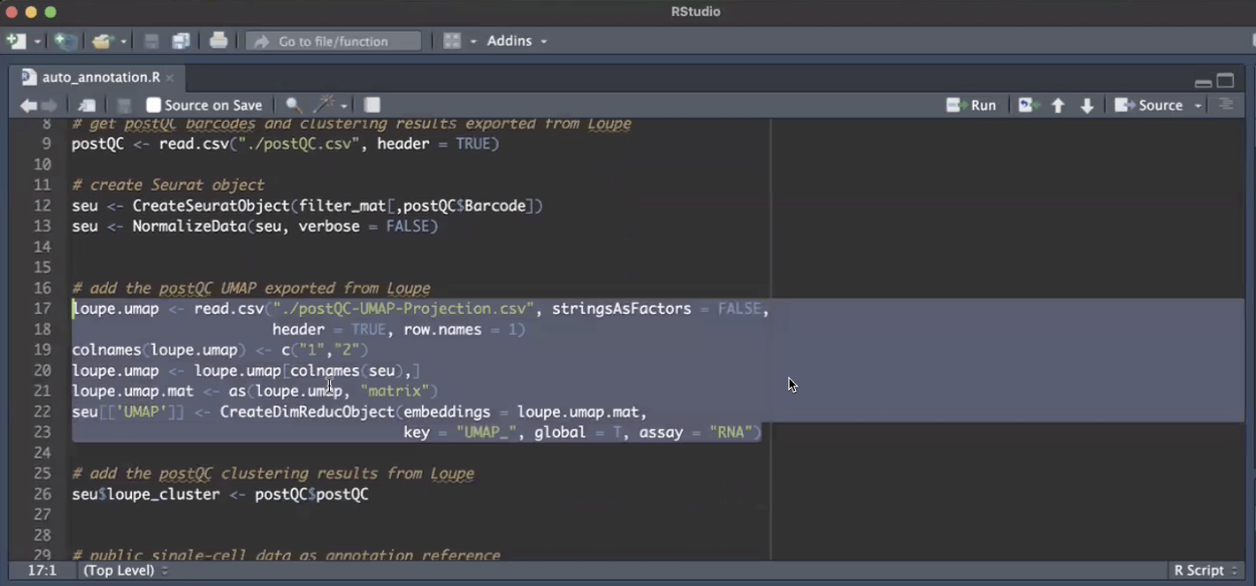
- 利用Broad Institude的PBMC公共数据集进行自动的细胞注释,需要确保公共数据集的格式也是Seurat的文件格式,我们需要把这个文件下载在本地

- 之后,我们就可以开始准备用Seurat自带的LabelTransfer算法,自动地把公共数据集的single-cell数据注释转移到我们的single-cell上,就帮我们省去了自己找marker-解释marker的麻烦。
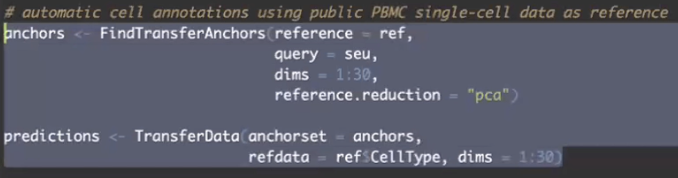
- 用LoupeR的软件包,将Seurat注释完的数据转换成Loupe Browser的文件,我们可以直接用Loupe Browser打开转换好的cloupe文件
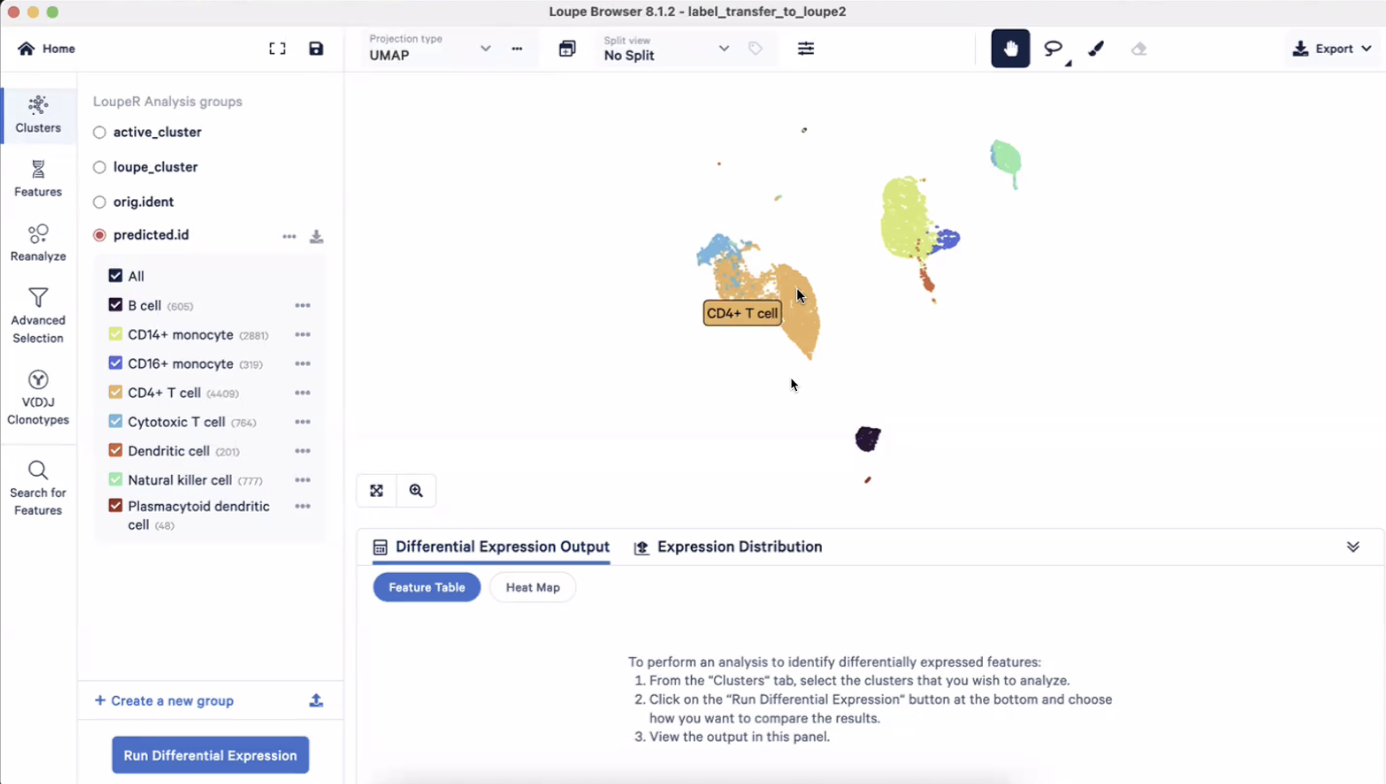
- 可以看到注释前后的数据,UMAP图是一样的,没有变化,下图展示的是经过自动注释后的UMAP图和clustering结果


- 每个细胞也已经被自动地注释为对应的细胞类型了(用Broad Institude的公共PBMC数据进行参考)
多样本的数据分析:pseudo-bulk
- 比如疾病组有3个样本,对照组有3个样本,这些样本中都有B细胞,我们想关注疾病组的B细胞与对照组的B细胞有哪些基因表达水平的差异
- 推荐:pseudo-bulk的多样本分析,在Loupe Browser中就可用
- 准备文件:含有(细胞barcode,donor(样本来源),condition(样本供者的状态)的csv文件,文件的内容如下图所示,需要包含headers

- 我们需要上传准备好的csv文件到Loupe Browser,之后就能看到不同的Group,比如这里我们可以看到donor和condition这两个分组group
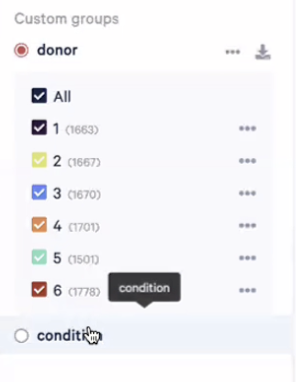
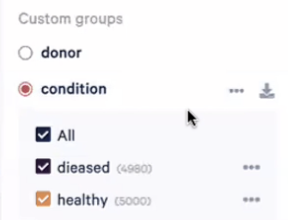
- 接下来,我们就可以准备好开始进行多样本的差异表达分析,比如如果我们想探究疾病组和对照组的B细胞有什么差异,我们可以点击app左下角的”Run Differential Expression”,然后在弹出来的对话框中选择”Across multiple samples”,然后”Start Analysis”
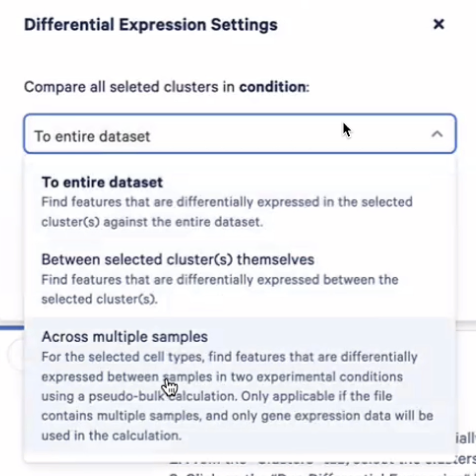
- 首先,我们要输入研究的细胞类型,这个信息在我们通过Seurat自动注释的细胞类型里面,也就是predicted.id,细胞类型选择B cell
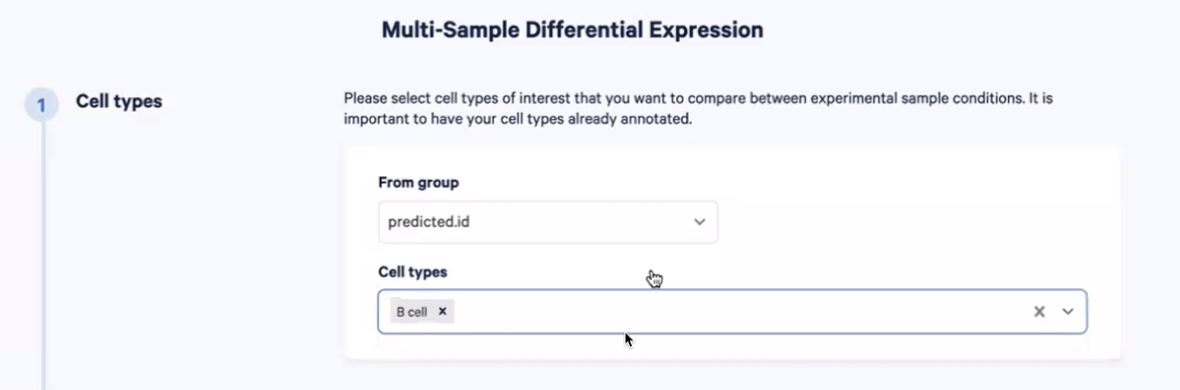
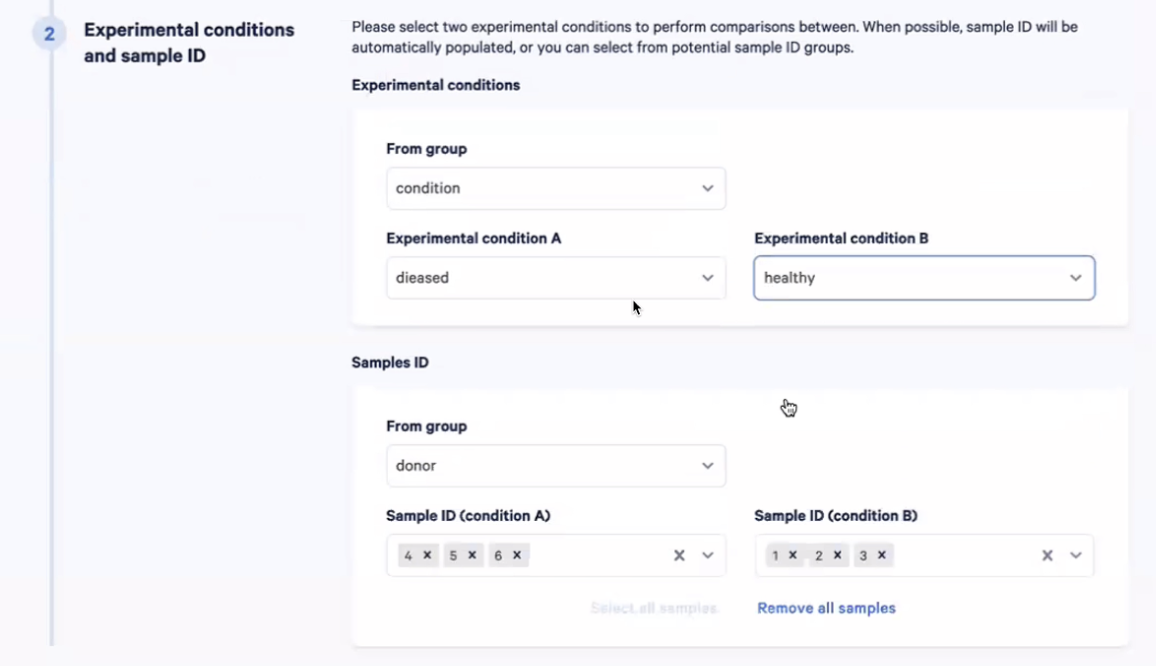
- 接着,我们回答我们要研究的细胞在哪两个组之间进行比较,我们可以在condition中获得这些细胞的donor是否健康的信息,我们还可以确定样本的ID,之后就可以开始计算了
观察样本的转录本捕获情况
这部分的内容是在解释为什么PBMC的样本会出现
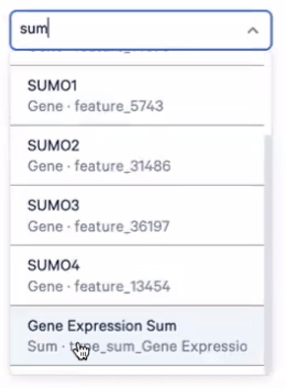
- 进入Features界面,在Search genes/features框中输入sum,然后选择Gene Expression Sum
- 这个时候,我们所看到的UMAP图展示的就是每个细胞的总的UMI数目了,可以看到淋系的细胞表达量会低一些,而髓系的细胞表达量会略高
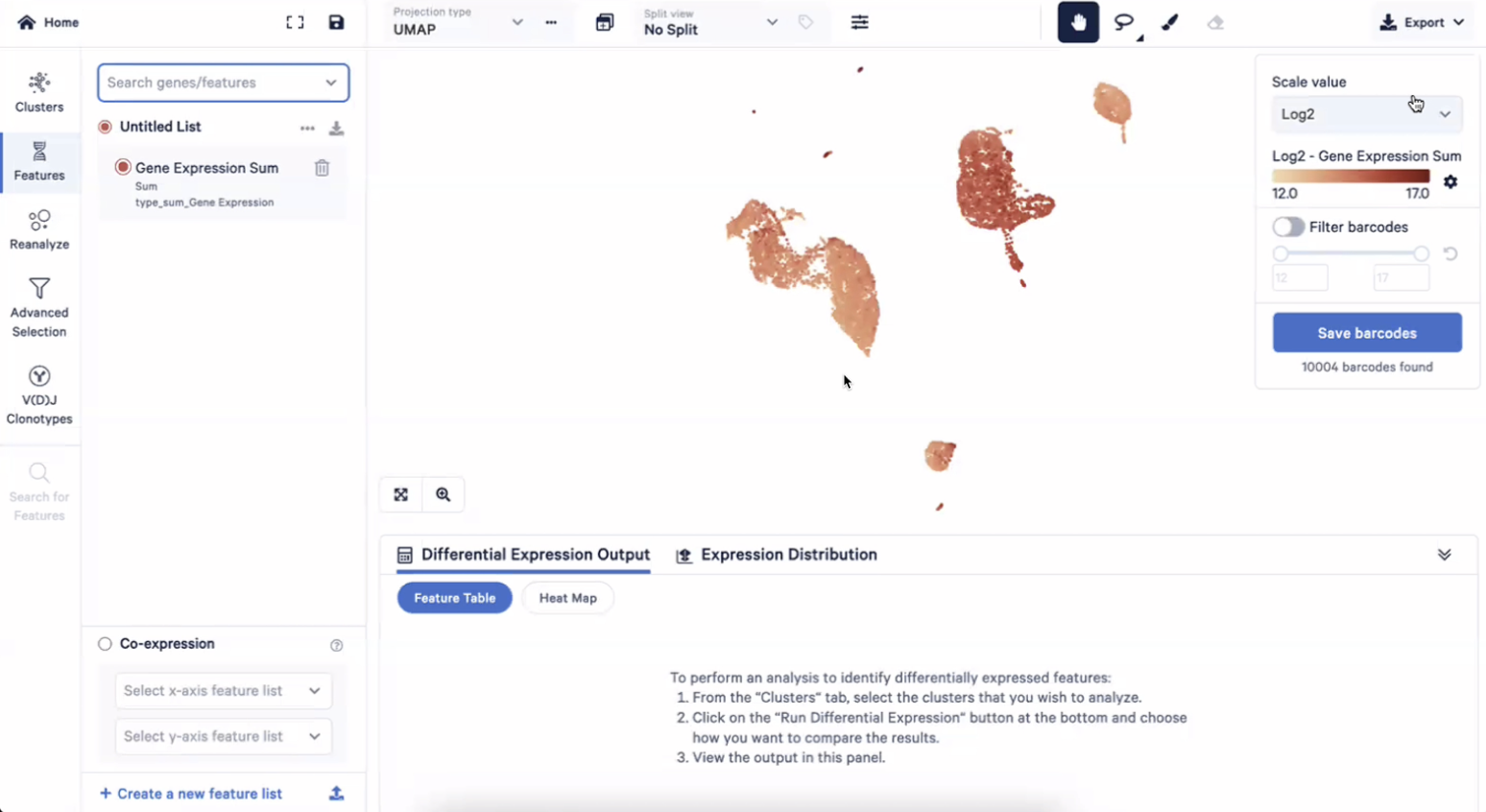
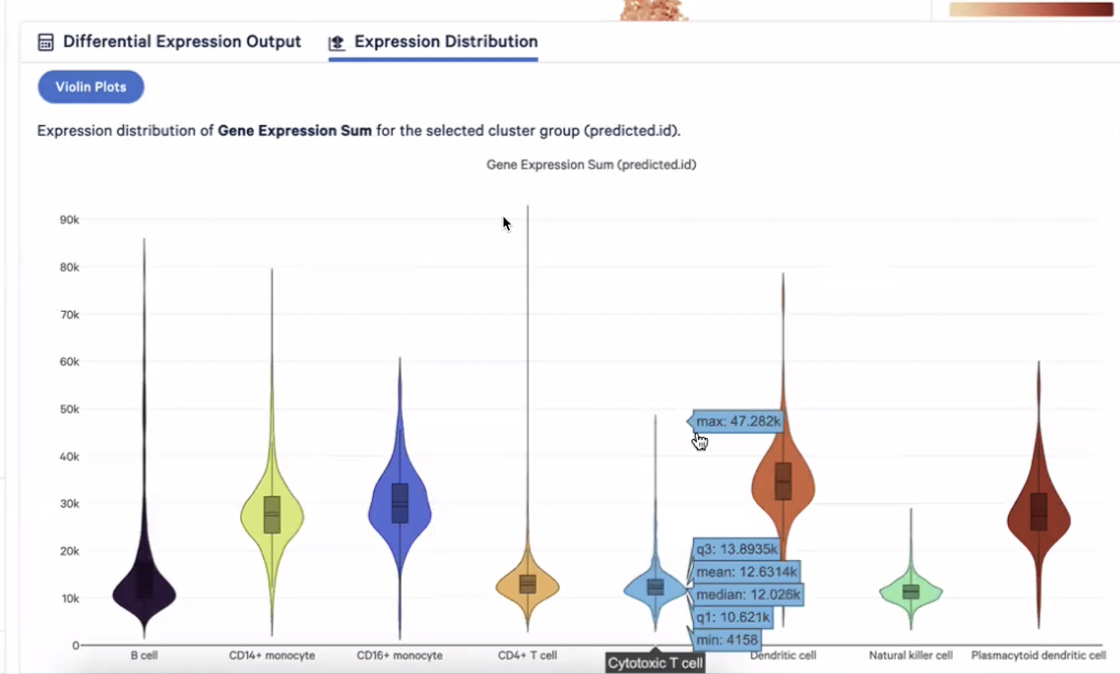
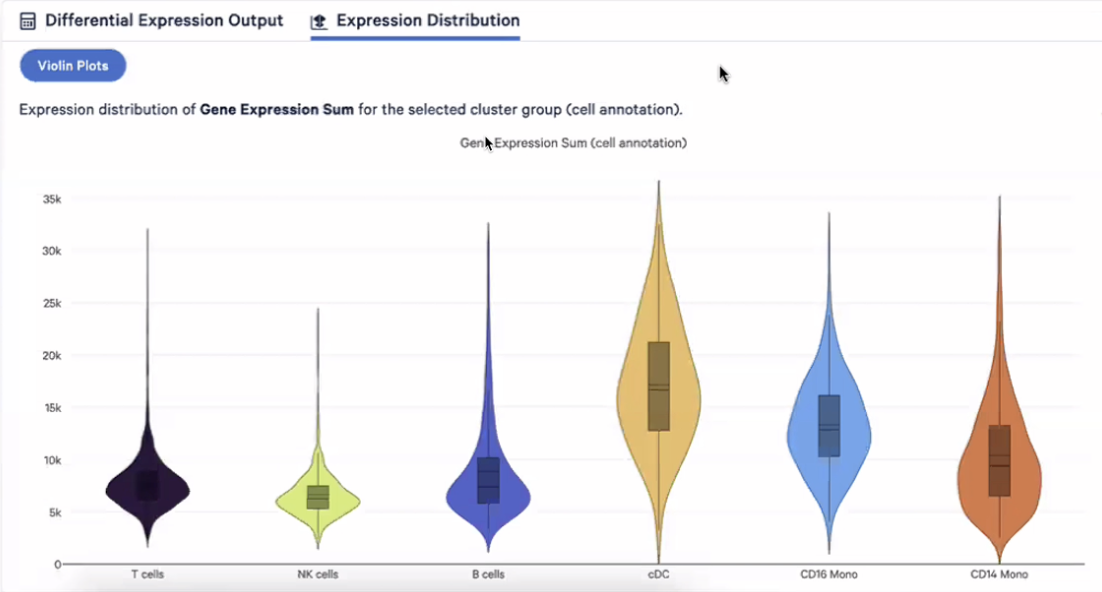
- 这样的双峰结构是由于GEM-X的捕获敏感度提高,在之前的技术中是看不到这样的结果的,下图展示了之前的技术的结果,分别是淋系7000左右,髓系9000左右
补充Q&A
如何判断测序是否饱和
- Cell Ranger:Web Summary文件可以给我们展示一个0-100%之间的测序饱和度值,和不同的测序深度对应的测序饱和度图,我们可以根据这两个指标来判断测序有没有到”sweet point”
Normalization
- 推荐在观察某个基因在不同的cluster中的表达情况时,去展示标准化算法之后的值,比如LogNorm,去除technical variation而保留biological variation
数据整合
- 在整合多个数据集进行分析的时候,有些数据集不是10x的标准文件格式,比如metadata和count文件,在整合分析时无法直接merge,对应的解决办法?
- 用R把count文件读成一个表格的形式,然后把表格转化成sparse matrix,sparse matrix可以被Seurat阅读,产生对应的Seurat文件,Metadata也可以转入到中Seurat
- 我们可以想办法先把多个样本的matrix放到一个Seurat数据里面,然后通过LoupeR把Seurat数据转换为Loupe Browser文件
不要乱用线粒体基因百分比去QC
- 不要看到Seurat教程中把5%线粒体基因作为上限,就照搬到自己的细胞上去,这个教程中用的是PBMC的数据,线粒体基因在这类细胞里面表达的比例本身就低
- 还是要根据自己的数据中,线粒体基因百分比的分布情况进行分析,然后根据自己的情况取上限值
- 如果遇到UMI和线粒体基因百分比都偏高的细胞,我们对这样的细胞不建议去除,因为这些细胞的质量是OK的,只是它们可能需要大量进行呼吸作用
设备管理
- 对一个project使用一台电脑,一个Seurat版本,让所有软件包的版本都统一
核糖体RNA做QC
- 做是有意义的,但是不做也不一定会被审稿人argue