数据分析概述
- 和其他的实验过程一样,10x推荐对空间实验的流程以及数据分析预先进行仔细的设计,从最开始的科学问题出发,详细设计空间实验的流程并产生数据。
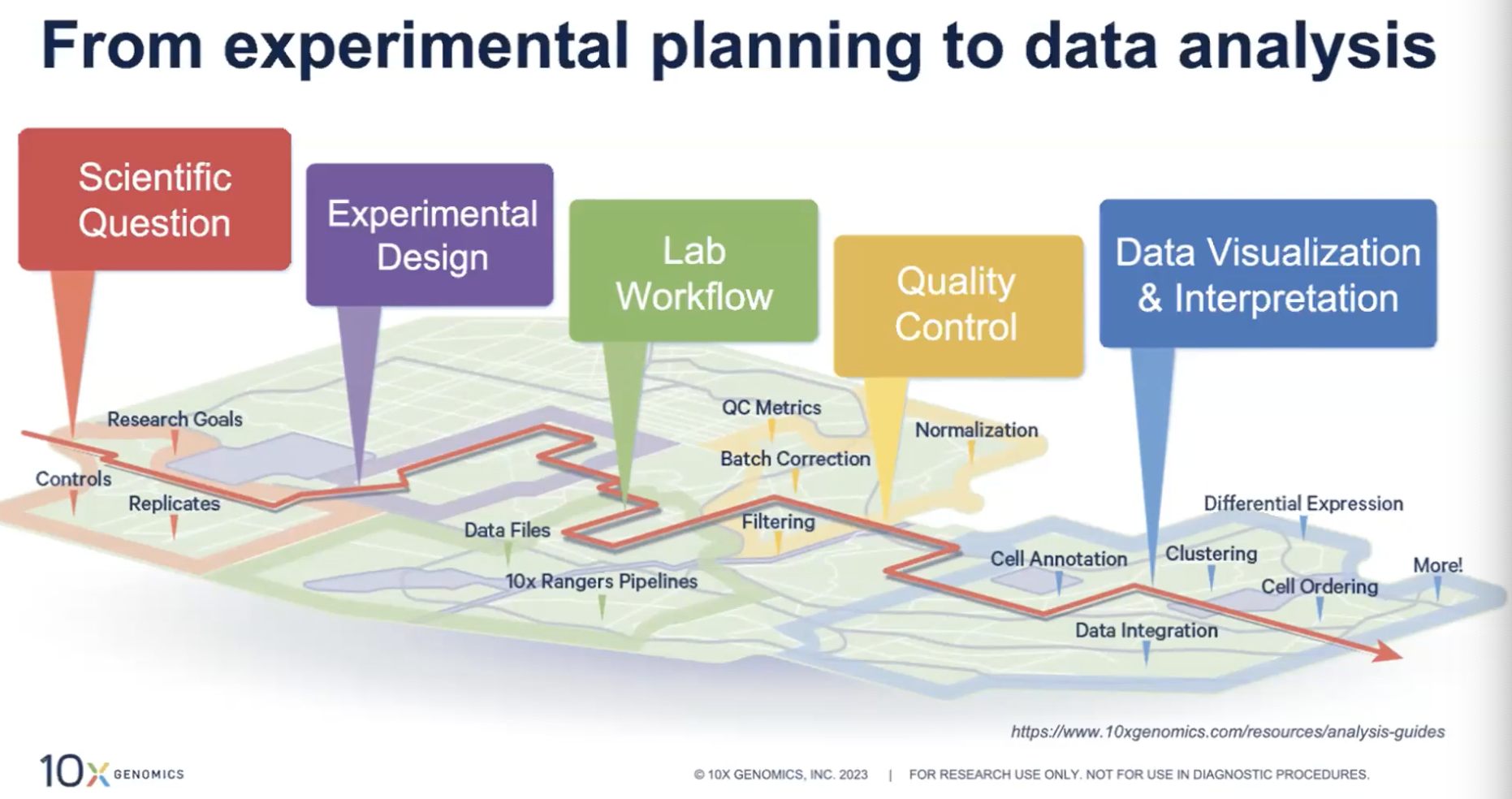
- 走到数据分析这一步时,基于课题的目的,需要设计数据分析的流程和步骤是什么,包括如何对数据进行质量控制QC,如何可视化数据,有什么样的分析流程需要进行,哪些软件工具可以实现这些分析目的,最后如何解读这些数据分析的结果。
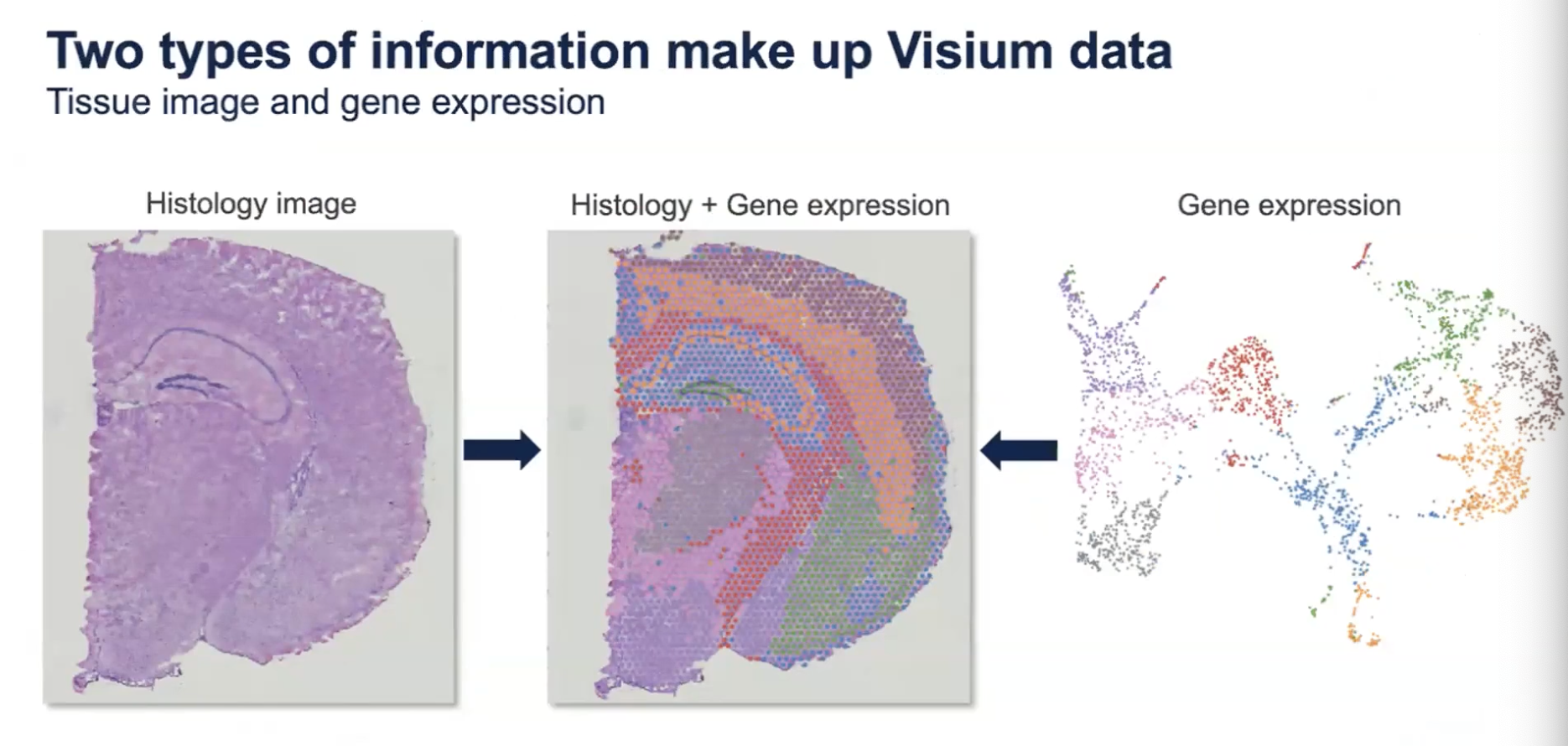
- Visium数据一般来说是由两种类型组成,分别是显微镜下得到的高清晰度的组织学切片图,以及用于测量基因表达的测序数据。Visium数据分析的强大之处在于如何去整合这两类数据。
- 可以从组织学切片开始,通过识别感兴趣的组织区域,然后叠加基因表达数据以寻找感兴趣的组织区域中的差异基因表达特征或者标记基因;也可以从基因表达数据开始,查看感兴趣的基因的表达情况,并探索聚类结果,以寻找有趣的基因表达特征,然后添加组织信息,并查看感兴趣的基因和聚类结果在组织中的位置。
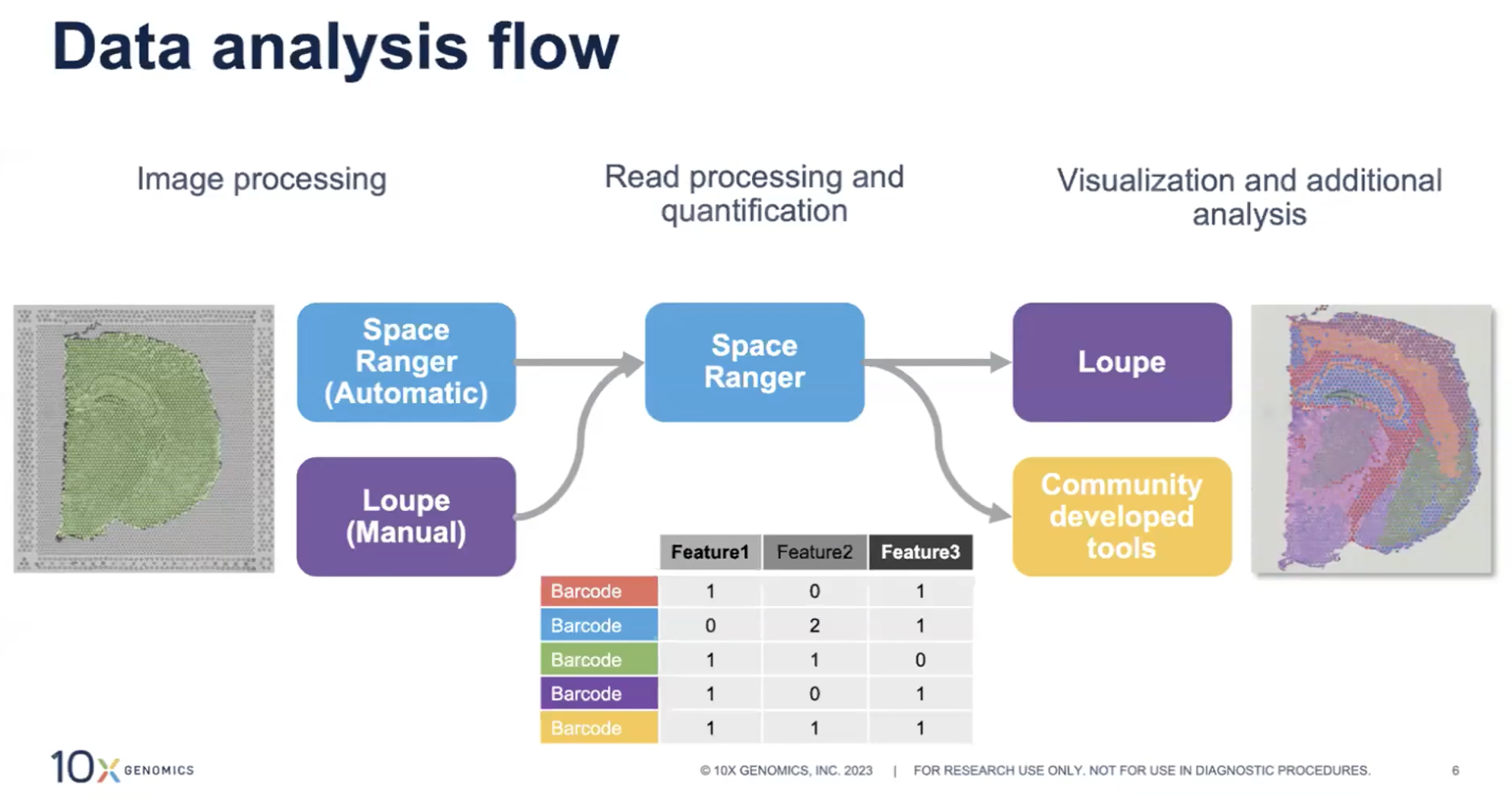
- 在Visium空间的数据分析中,需要处理两类数据,分别是Visium图片和测序数据的结果。一般来说,空间数据分析的流程包括对图片的处理、测序数据read的处理和基因表达量化,数据的可视化和下游数据分析。
- 首先,Visium图片首先会被处理,以寻找spot相对组织切片的位置,并找到覆盖组织区域的spot,这个过程可以用10X研发的SpaceRanger全程自动进行,SpaceRanger对图片的自动对齐算法可以实现图片的自动处理,但是由于SpaceRanger自动对齐的算法并非十全十美,有些时候如果图片质量不是特别好,自动对齐结果可能就不是很理想,在这种情况下可以使用10X研发的Loupe Browser进行手动对齐,确定spot的位置,并找到覆盖组织区域的spot。
- 完成第一步之后,SpaceRanger会进一步的对测序数据进行处理,并计算出每一个spot里面的基因表达量,从而得到基因和spot的矩阵。
- 最后,可以对覆盖组织区域的spot基因表达量进行可视化,以及额外的下游数据分析,包括QC,聚类,差异基因表达分析,以及spot的反卷积spot decovolution等等,可以用10x官方的的Loupe Browser或者是第三方软件实现这些不同的下游数据分析。
应用到数据分析中的软件

工具简介
SpaceRanger
- SpaceRanger是一个处理10X空间Visium数据的工具包,由10X团队研发;SpaceRanger可以直接处理Visium的图片数据和测序数据,以此获得覆盖组织区域的spot的基因表达情况。
Loupe Browser
- Loupe也是10X团队研发出来的,是一个免费的桌面工具,用于空间Visium数据的可视化和分析。下文会展示Loupe Browser的功能界面,并展示如何用Loupe来分析的空间Visium数据。
- 纵使的Loupe已经拥有很多的功能,但Loupe能够提供的分析方法毕竟有限;如果想要进行更高阶的数据分析,比如去批次效应、Spot Deconvolution等,还是需要使用第三方软件;这些三方软件主要是一些独立运行的软件包,包括大家熟知的Seurat,它们大部分是由全世界不同的科研团队研发的。
- 这些软件非常有用,但由于这些软件不是10X研发的,10x对这些软件不能提供官方的技术支持;如果在这些软件的使用过程中遇到问题,10x会强烈推荐直接联系这些软件的开发团队。
SpaceRanger
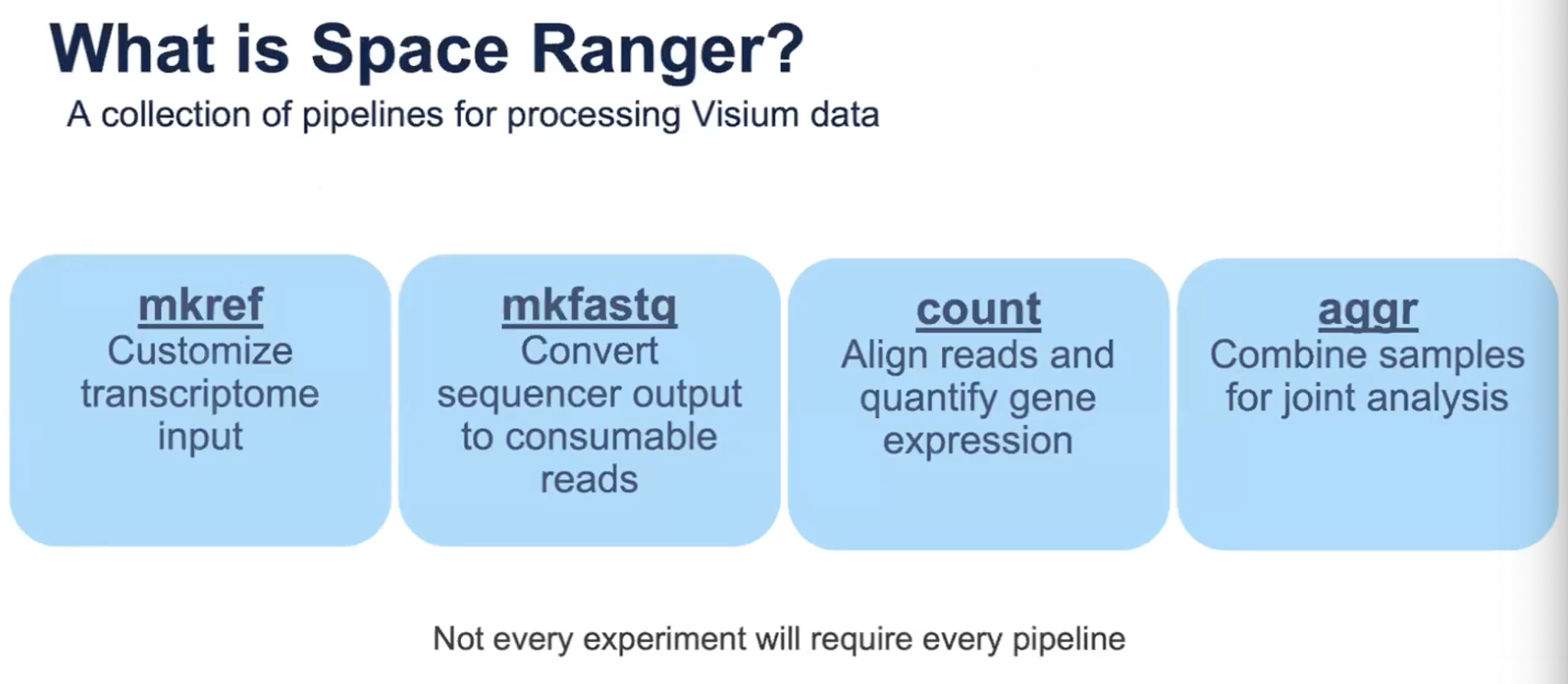
- SpaceRanger是应用于10x Visium数据的软件包,这个软件包有多个程序,包括mkref、mkfastq、count、aggr
- mkref:如果使用了Visium的版本1(Visium V1),新鲜冰冻样本,并且数据来源不是人类和小鼠,就需要SpaceRanger的makeref pipeline来定制参考基因组
- makefastq:更多被服务商或者是实验中心使用,作用是将测序后下机的bcl文件转换为fastq文件,作为终端用户拿到手的往往是转换好的fastq文件
- count:将Visium的图片和测序结果一起进行整合分析,产生重要的Spot和基因的矩阵,并跟组织图片进行结合。此外,如果实验设计中有多个样本,那就可以考虑使用aggr程序将不同的样本进行整合。
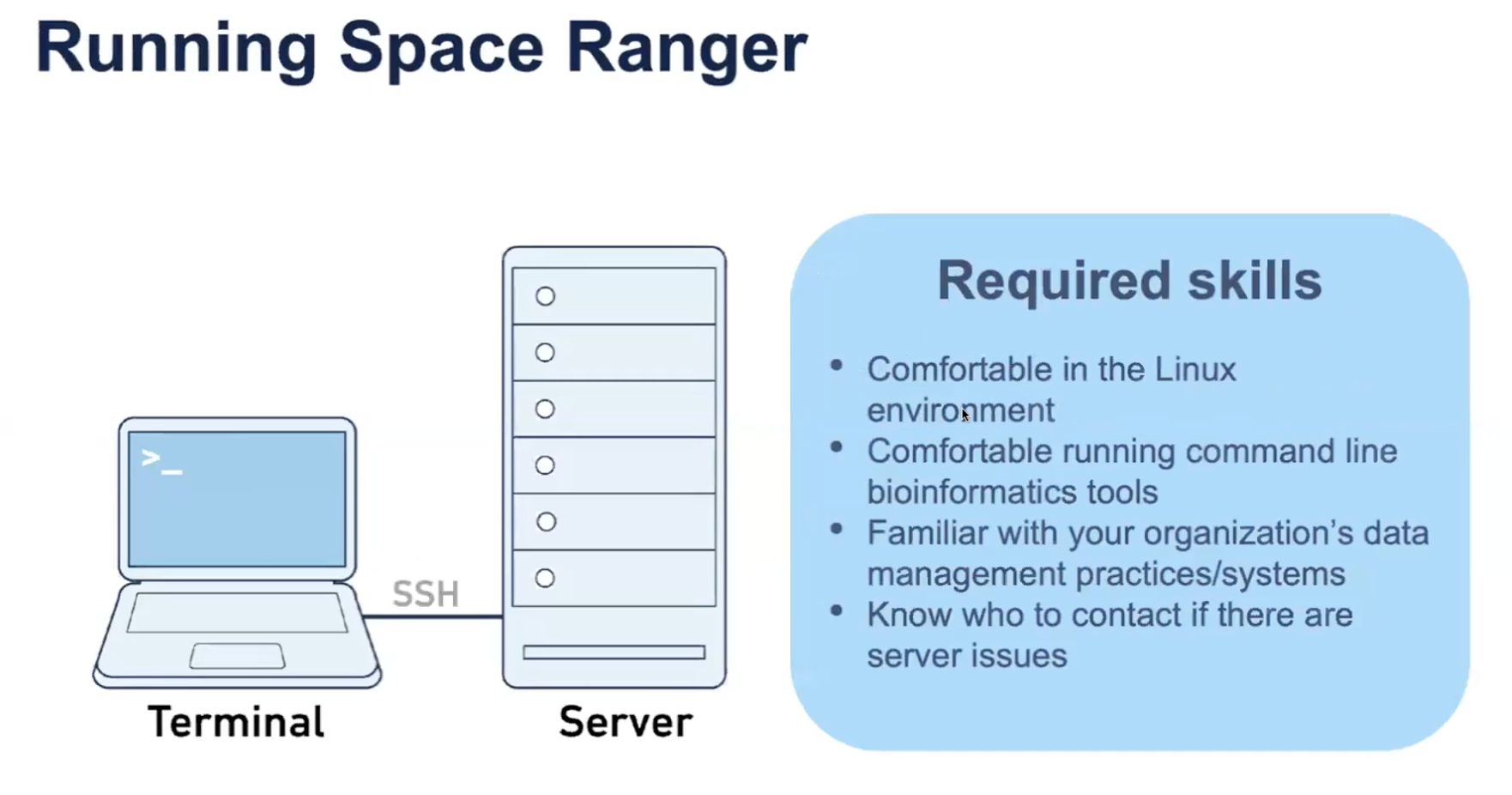
- SpaceRanger的运行要求
- Linux的运行环境
- 了解并熟悉运行命令行
- 了解所在实验室或者研究所的服务器系统
- 如果出现了服务器相关的问题,该去联系谁
Loupe Browser
- Loupe Browser是10x开发的桌面软件,可以可视化和分析10x的数据
- 它非常简单易上手,友好的用户界面适用于任何使用人员,不需要学习任何的编程语言就可以使用Loupe Browser

- Loupe Browser的使用要求
- 熟练使用苹果或微软的操作系统
- 了解生物学和实验的相关知识
- 了解实验设计和数据表达的结果
第三方软件
- 除了10x研发的Space Ranger和Loupe Browser之外,其他科研团队也研发了大量第三方软件,同样支持Visium的数据分析;这些软件通常是学术界不同的科研团队独立研发出的软件工具,这些工具可以为不同的数据分析目的服务。
- 举例:一般来说,Visium的Spot直径在55微米左右,大部分情况下会覆盖2到4个细胞,为了能够了解每一个Spot里面细胞类型的组成部分,可能需要需要一些第三方软件实现反卷积(英文spot deconvolution);无论是否有单细胞数据,都可以用不同的软件实现Spot Deconvolution
- 如果有多个样本想要进行数据整合,就要考虑批次效应在样本间的影响;这时,一些三方软件比如Seurat或Harmony可以提供有效的批次矫正;另一些软件,比如SpaceGCN可以整合空间信息,以基因表达数据进行聚类分析,从而实现所说的空间聚类分析,并识别出空间上的基因表达差异。
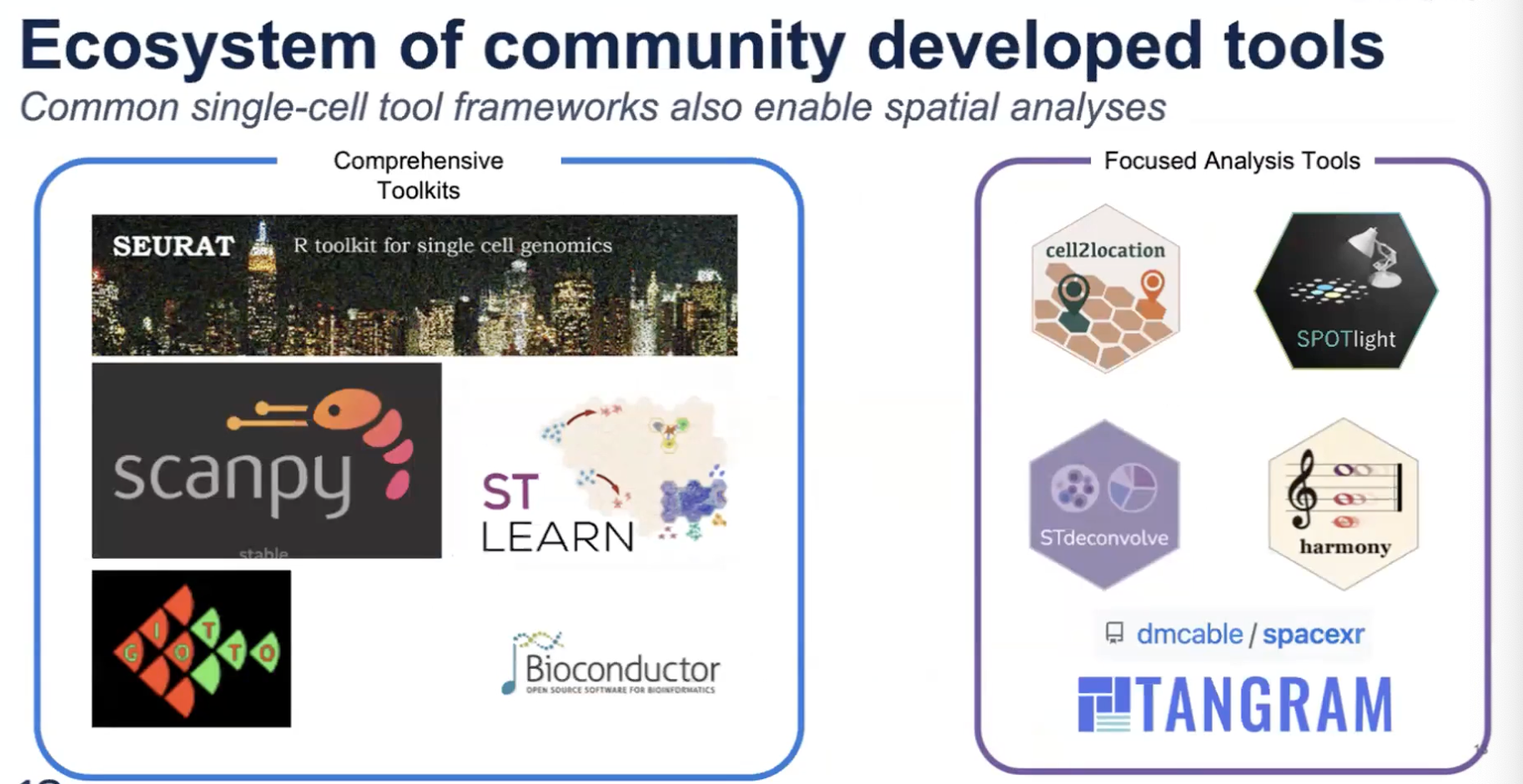
- 三方软件可以分成两个部分,分别是上图左侧的综合分析软件和上图右侧的专项分析软件
- 很多用于单细胞转录组数据分析的软件同样可以运用在空间数据中,比如Seurat、scanpy和harmony
- 综合分析软件提供的程序可以覆盖Visium数据分析中的不同步骤,包括QC、降维、聚类分析、t-SNE和UMAP投射,以及空间的数据可视化。此外,这些软件也提供一些特殊的分析方法,比如STLEARN可以提供空间的聚类、空间细胞的相互作用以及空间中的trajectory分析。
- 专项分析软件可以提供基于特定目的的数据分析,比如harmony可以用于进行批次矫正, Cell2Location可以将单细胞数据和Visium数据进行整合,进行基于单细胞数据的Spot Deconvolution,而STdeconvolve可以对Visium数据直接进行Spot Deconvolution,不需要单细胞数据的参与。
- 这些软件极大拓宽了Visium数据分析的广度和深度,然而由于这些软件并非是由10x研发的,因此没有正式的官方技术支持
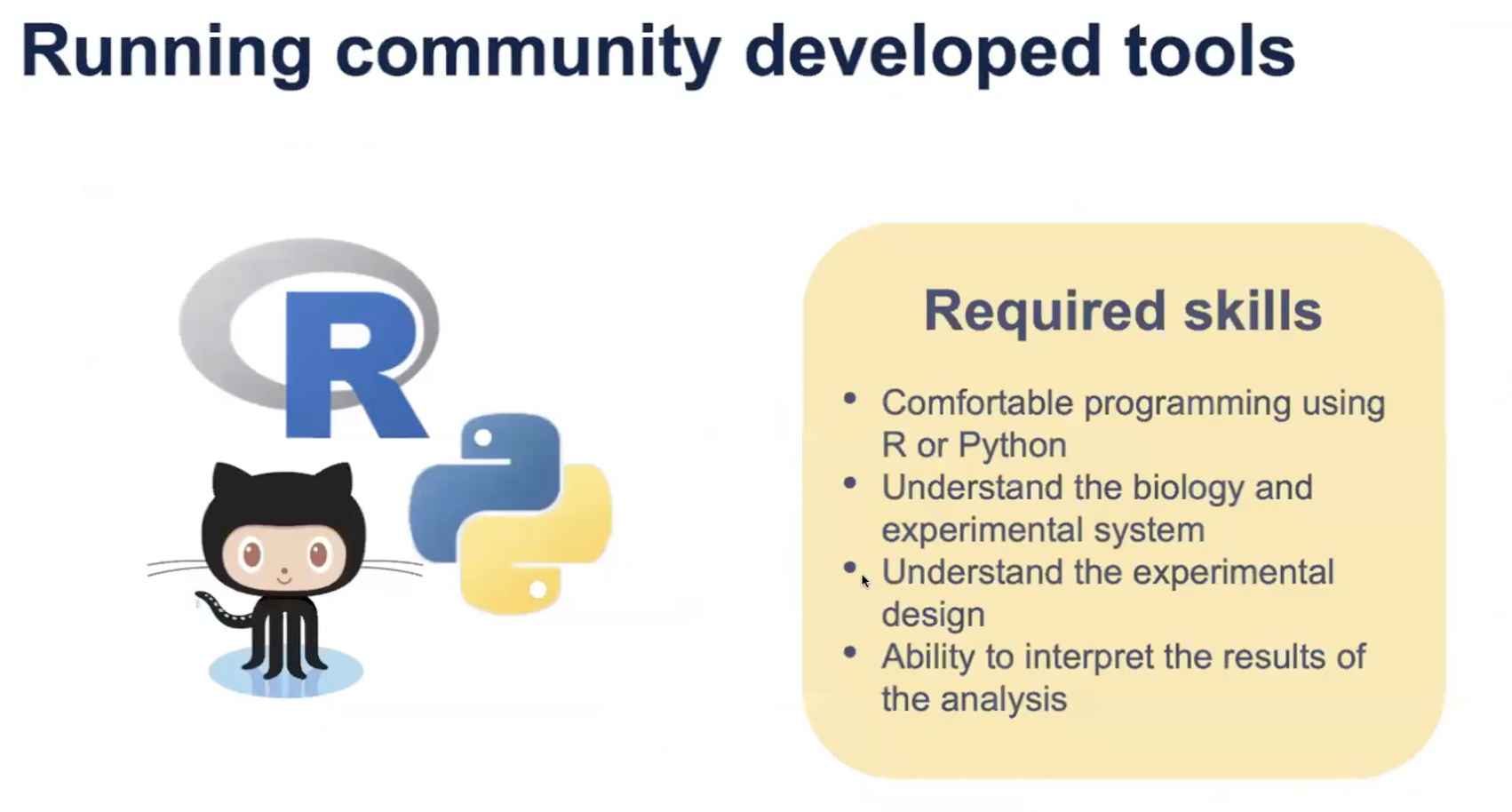
- 三方软件的使用要求
- 了解相关的编程语言,比如说R或Python语言
- 了解生物学和实验的相关知识
- 了解相关的实验设计
- 解读数据分析的结果
Spot Deconvolution
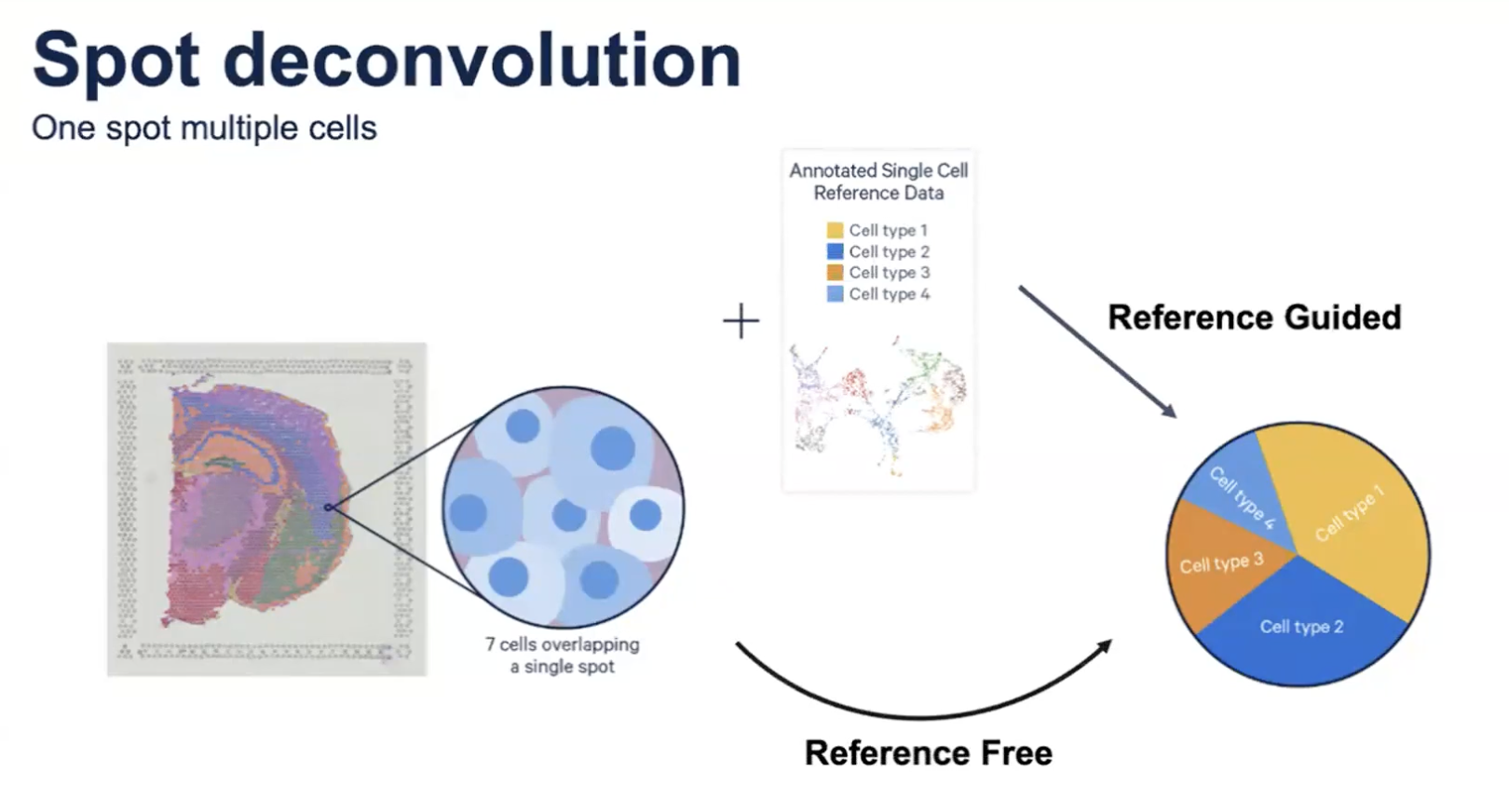
- 由于Visium的Spot直径在55微米,因此每个Spot大部分情况下会覆盖着2到4个细胞;如果想要了解某一个Spot里面的细胞组成,那么有两种方法可以进行Spot Deconvolution,具体取决于是否有匹配的单细胞数据。
- 如果可以提供来自匹配的组织类型的单细胞数据,那就可以将单细胞和Visium数据进行结合,进行Spot Deconvolution的分析;如果没有组织类型匹配的单细胞数据,同样可以进行没有单细胞数据参与的Spot Deconvolution,称之为Reference Free Spot Deconvolution,这两种方法都可以破译Visium Spot里面的细胞组成类型。
- 需要特别提出的是,Space Ranger 2.1版本起已经支持没有单细胞数据参与的Spot Deconvolution
进行spot deconvolution的工具
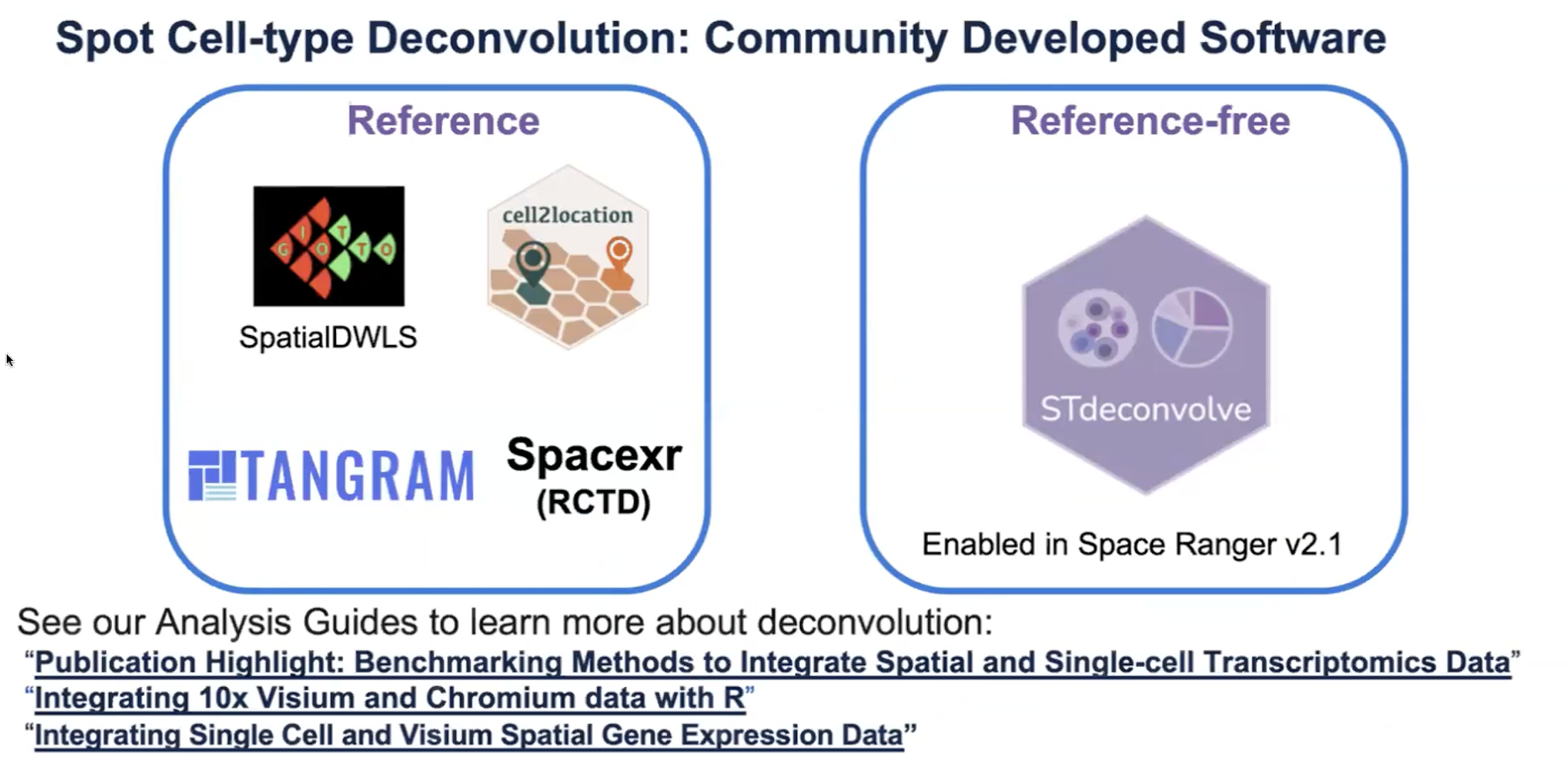
- 如果能提供匹配的单细胞数据,一些有用的三方软件可以将单细胞数据和Visium数据进行整合,例如cell2location和RCTD等,10x官网有分享了分析指南资源,提供了几篇有用的文章,比如说目前有超过15个工具,上图列出的第一篇文章就重点介绍了一些Benchmarking的测试工作,推荐了比较好用的Spot Deconvolution的软件,也有更多的文章推荐其他的三方软件对单细胞和Visium数据进行整合来进行Spot Deconvolution
- 但也有另外一种情况,如果没有匹配的单细胞数据,并且也想要做Spot Deconvolution,使用另一些软件工具也可以达到,比如STdeconvolve。此外,从SpaceRanger 2.1起,软件已经内置了跟ST Deconvolve非常相似的算法,可以自动进行没有单细胞数据参与的Spot Deconvolution。默认情况下,SpaceRanger的版本如果≥2.1,这个软件就会自动产生Reference Free Spot Deconvolution的结果,算法基于Latent Dilution类的Allocation模型去进行Spot Deconvolution。
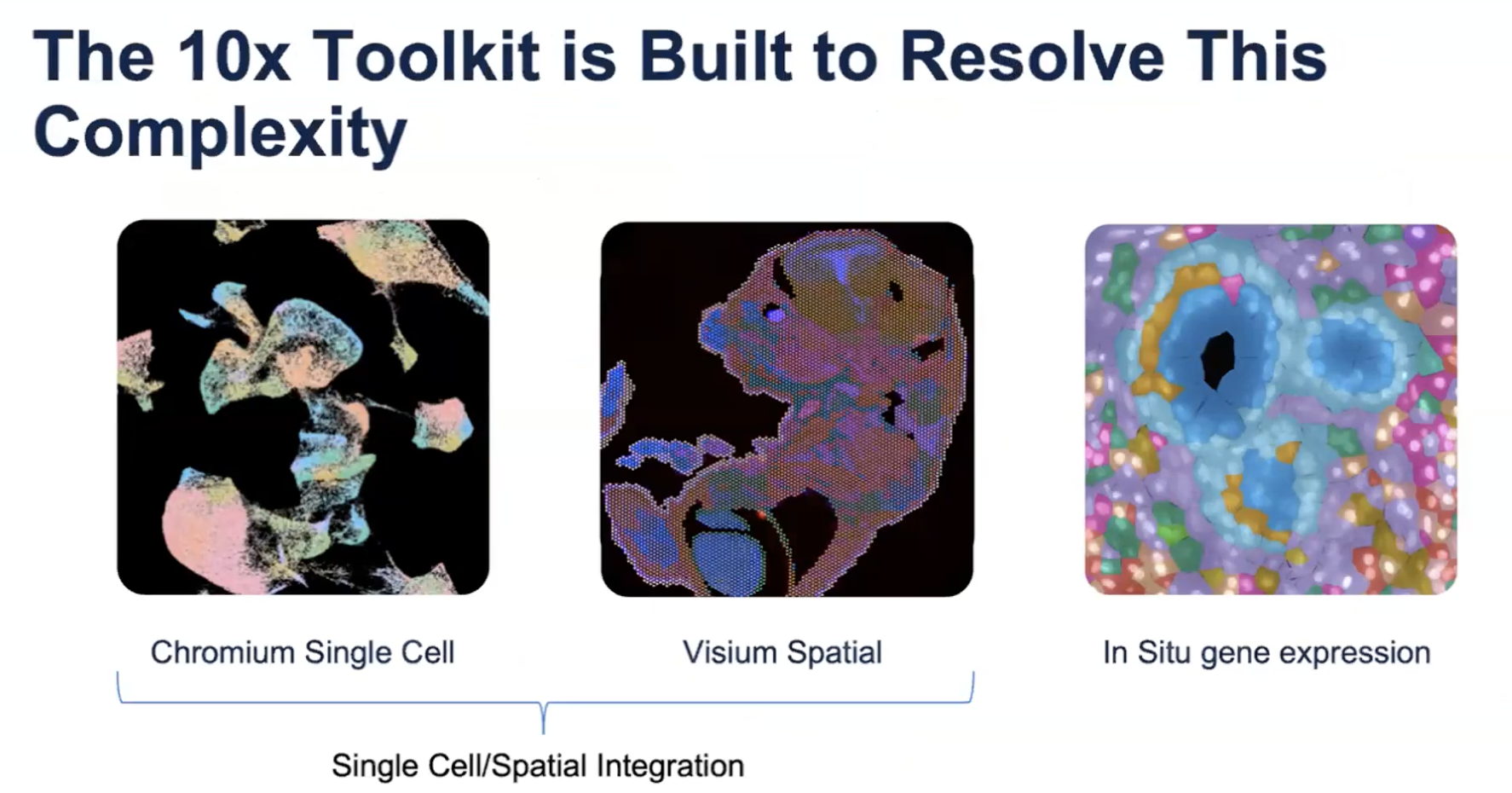

- 如果密切关注10x的产品目录,我们可以发现10x是唯一一个提供单细胞、空间Visium和原位Xenium三大平台的公司,现在单细胞和Visium的数据可以进行的有机的整合,从而了解在空间背景下每个细胞的基因表达情况。正是这种多数据整合,帮助了我们得到更多的生物学的发现,并解答一些重要的生物学问题,比如关注的基因在空间上的表达情况,这些基因的表达情况与潜在的病理学特征是否相关;关注的细胞在空间上的分布情况;在不同的组织区域和实验条件下是否有着不同的细胞类型分布和细胞相互作用;通过细胞相互作用的研究是否可以优化我的靶向治疗
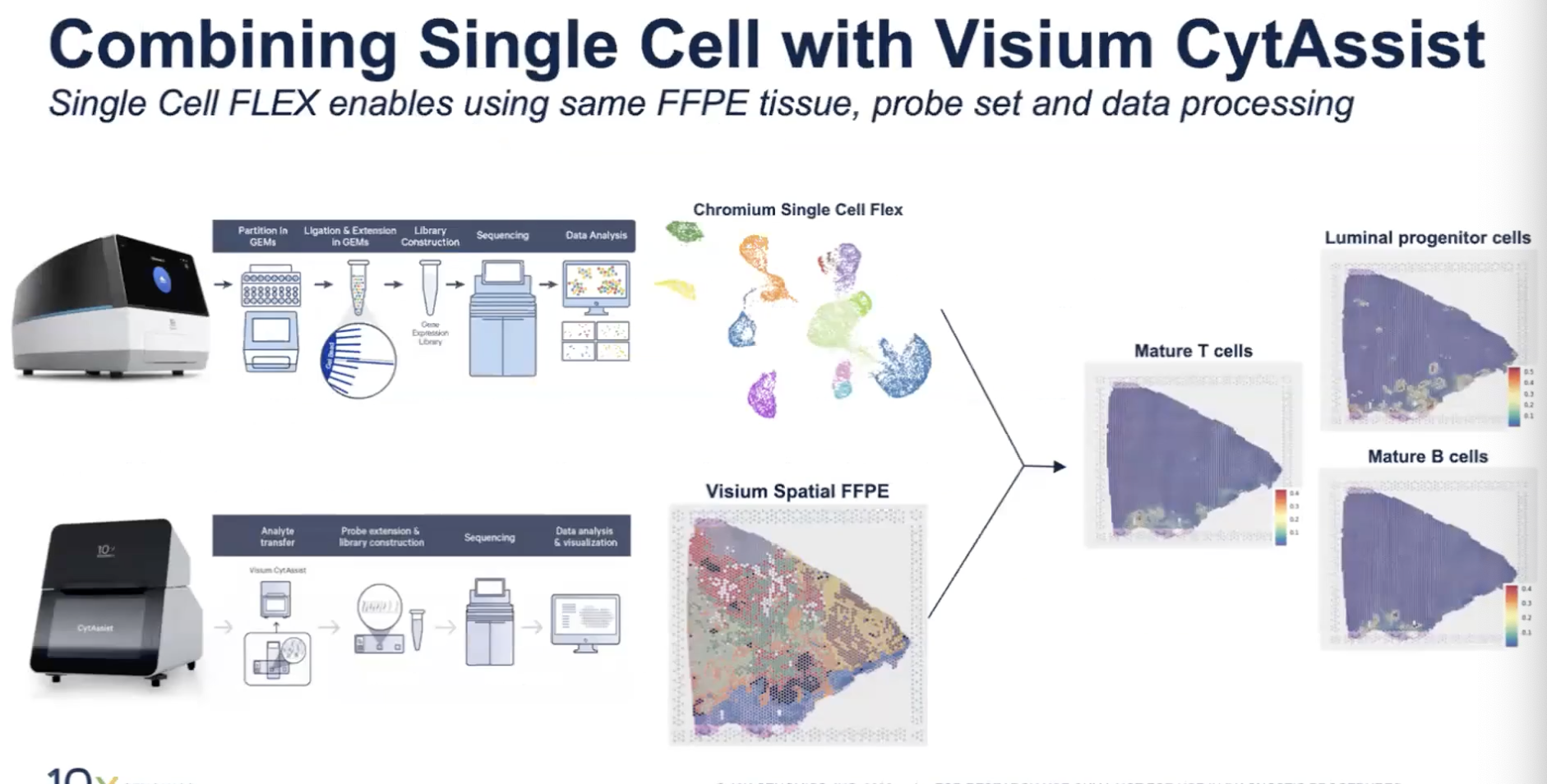
- 如果能提供病理学的FFPE组织库,我们就可以用单细胞的Flex和Visium Cylosis技术,在同一块FFPE组织块里面产生单细胞和空间数据,整合单细胞的Flex数据以及Visium Cylosis数据可以使我们更准确地在空间上定位感兴趣的细胞类型。
示例:人乳腺癌FFPE样本的Visium数据
- 当我们拿到Visium空间数据,包括的Visium图片和测序数据的时候,需要用Linux系统上的Space Ranger去处理这些图片数据和测序数据;Space Ranger运行成功后会产生一个非常重要的cloupe文件,这个cloupe文件可以直接导入Loupe Browser里面
- 今天的实例是人乳腺癌FFPE的样本,进行了Visium分析,并且同一个组织切片里也运用Flex技术进行了Single Cell的分析,两者整合后的结果可以帮助我们更好的了解这个乳腺癌的组织。
Loupe 7的全新界面
Spatial View
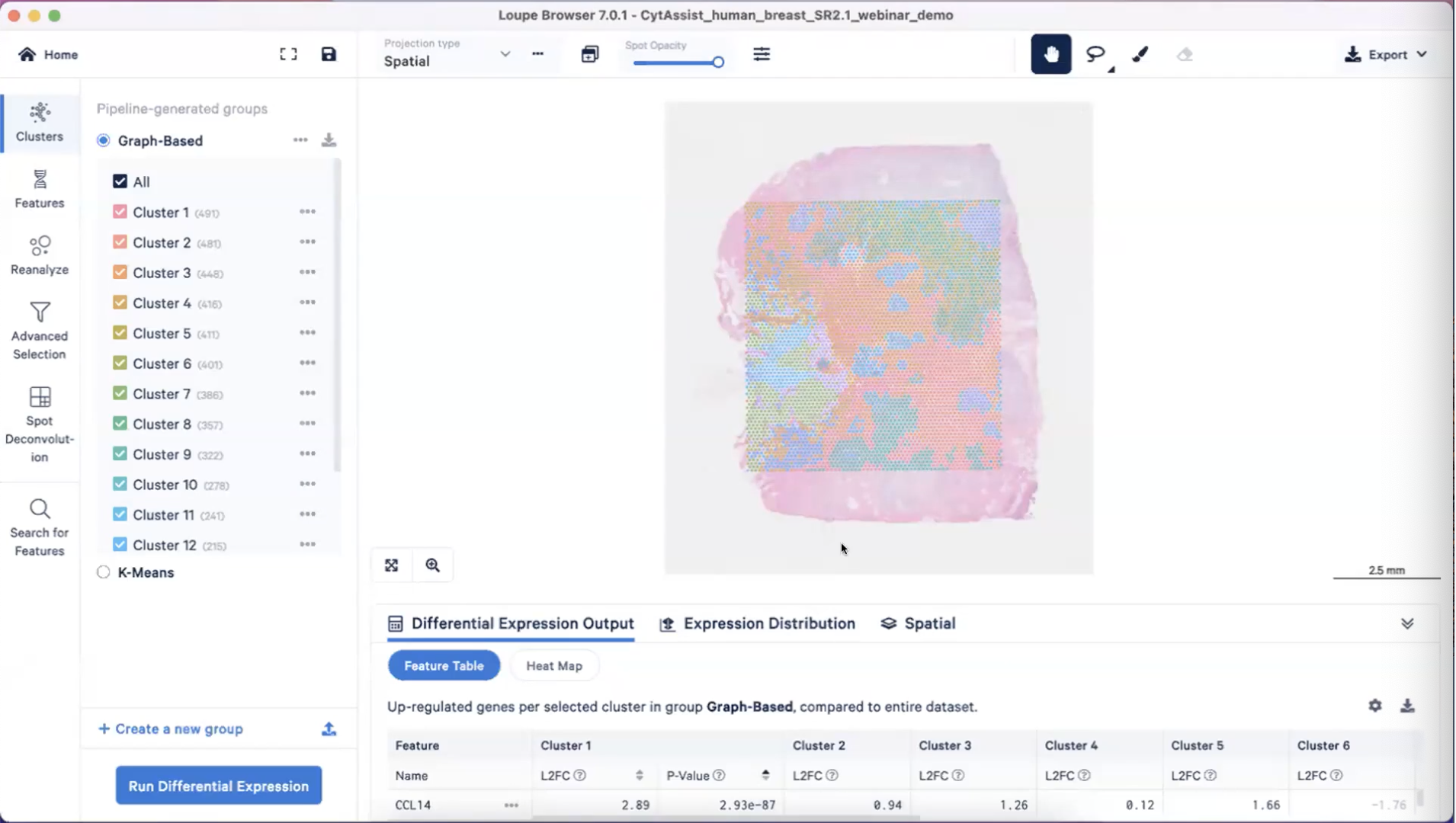
- Loupe 7的中间是Spatial View,Spatial View的底层是一个非常清晰的组织切片,上面覆盖着4992个Spot
t-SNE与UMAP
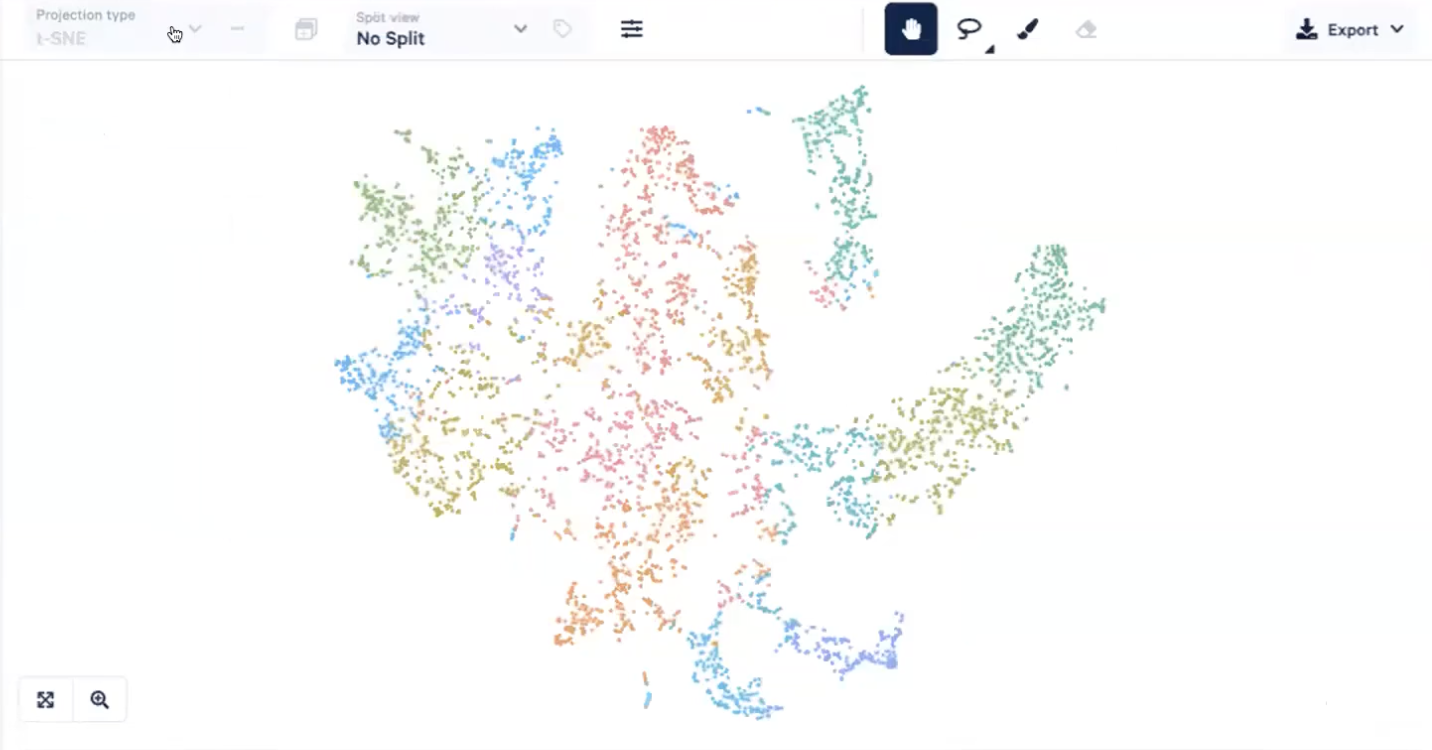
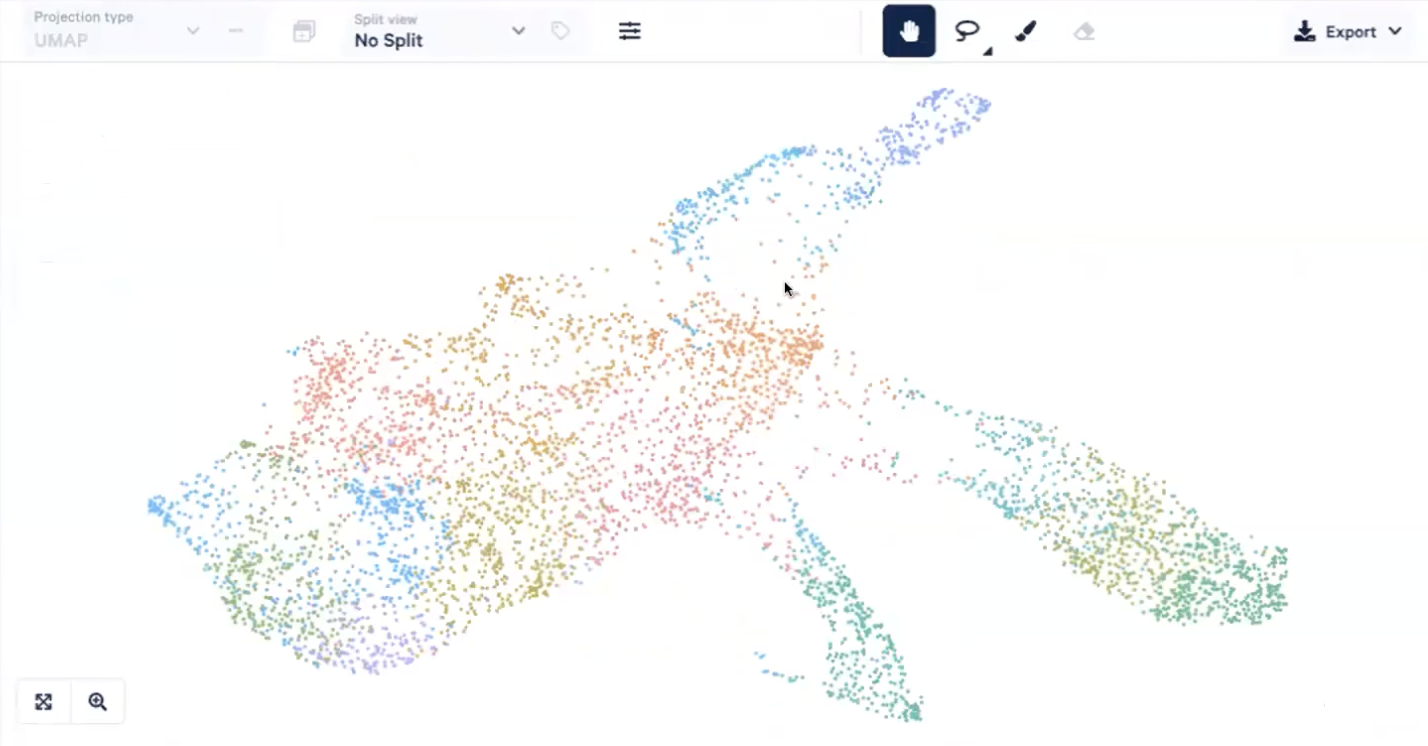
- 我们可以在”projection type”中选择t-SNE或UMAP,每个点就是一个Spot,不同颜色的点对应着不同的cluster结果
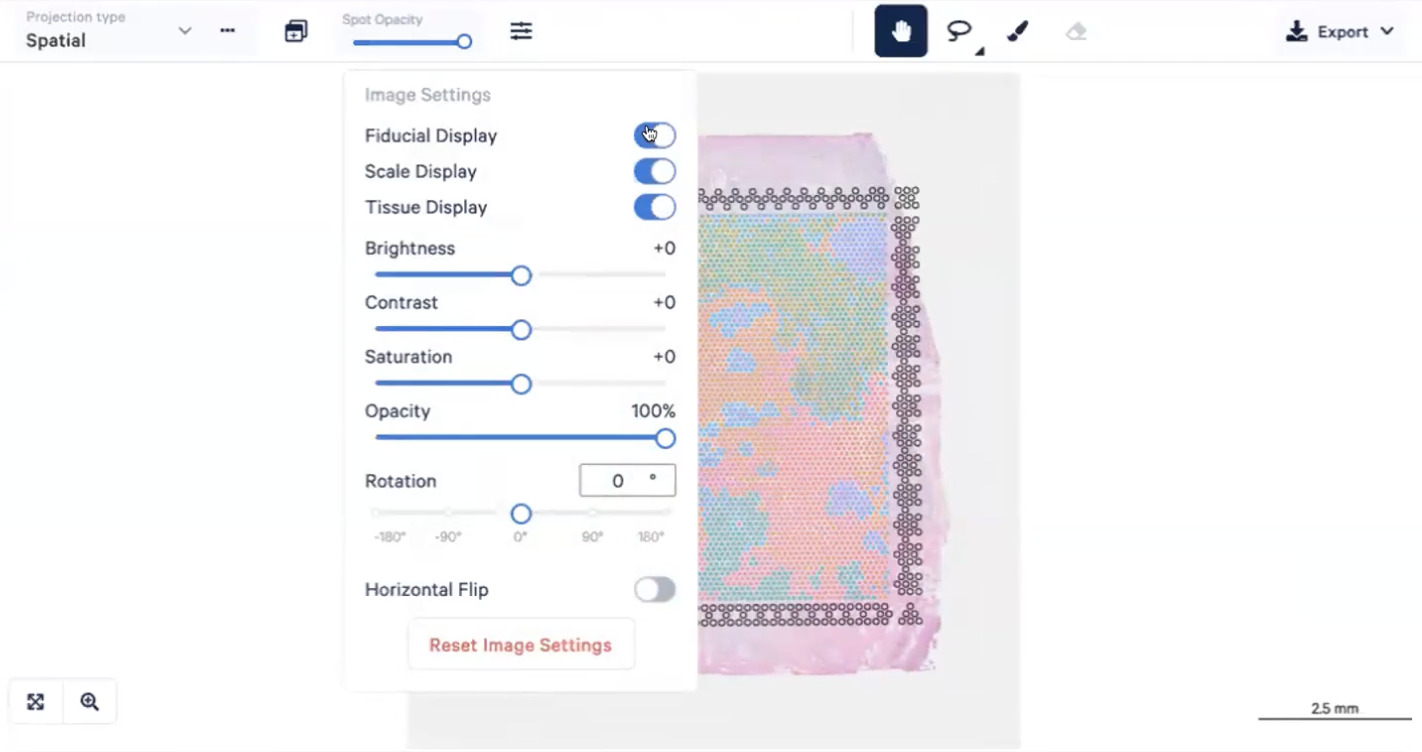
- 回到Spatial View,我们可以在Spot Opacity右侧的按钮进行图片的调整,比如可以显示用于确定位置的Fiducial Frame
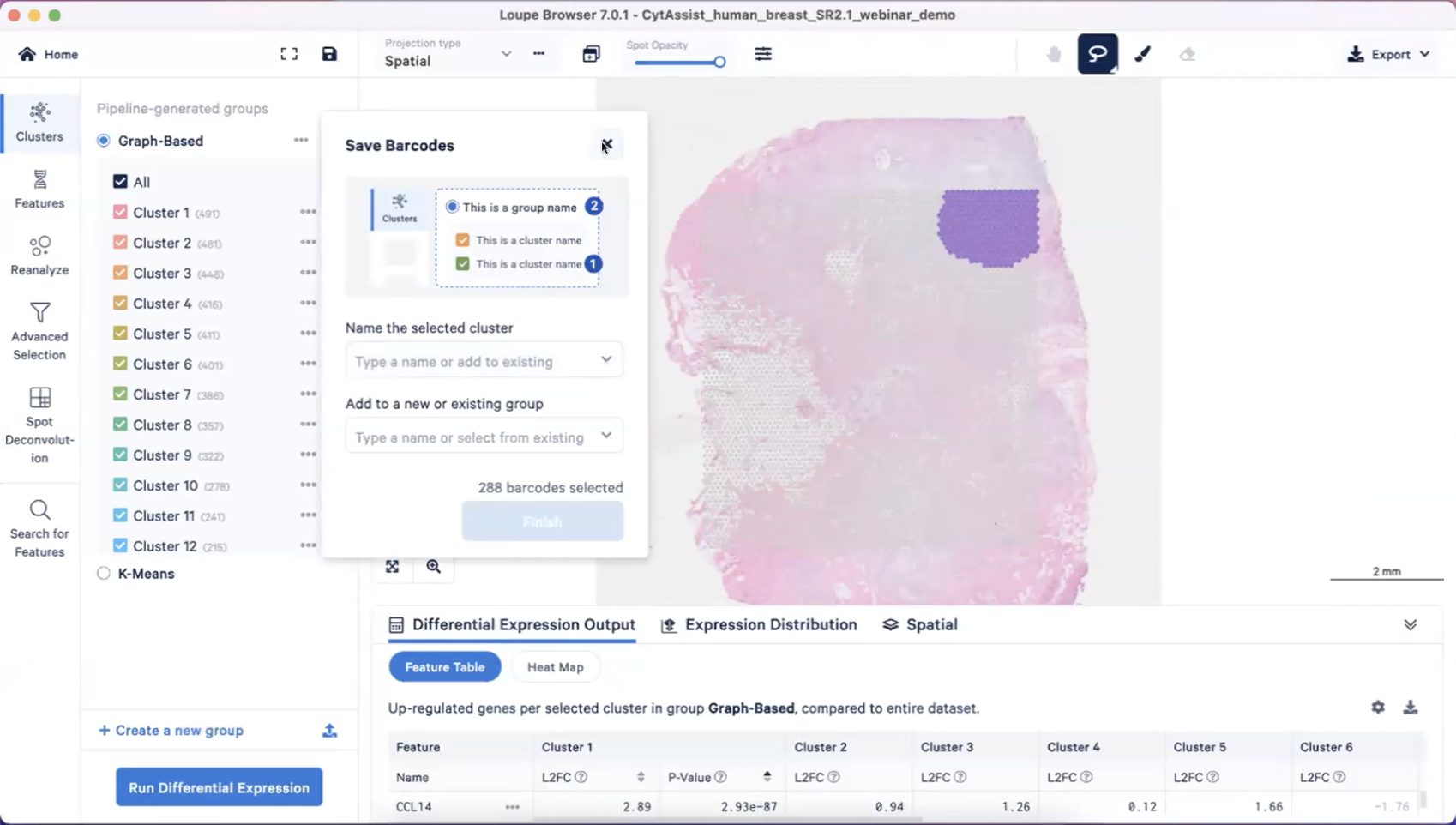
- 右上角的工具可以对spot进行选择,然后人工划分出我们感兴趣的区域,并把他们划归到同一个组上,就像下图这样
导出Spatial/t-SNE/UMAP的结果
- 可以使用右上角的Export按钮,将Spatial/t-SNE/UMAP图导出成PNG或SVG格式
clusters功能
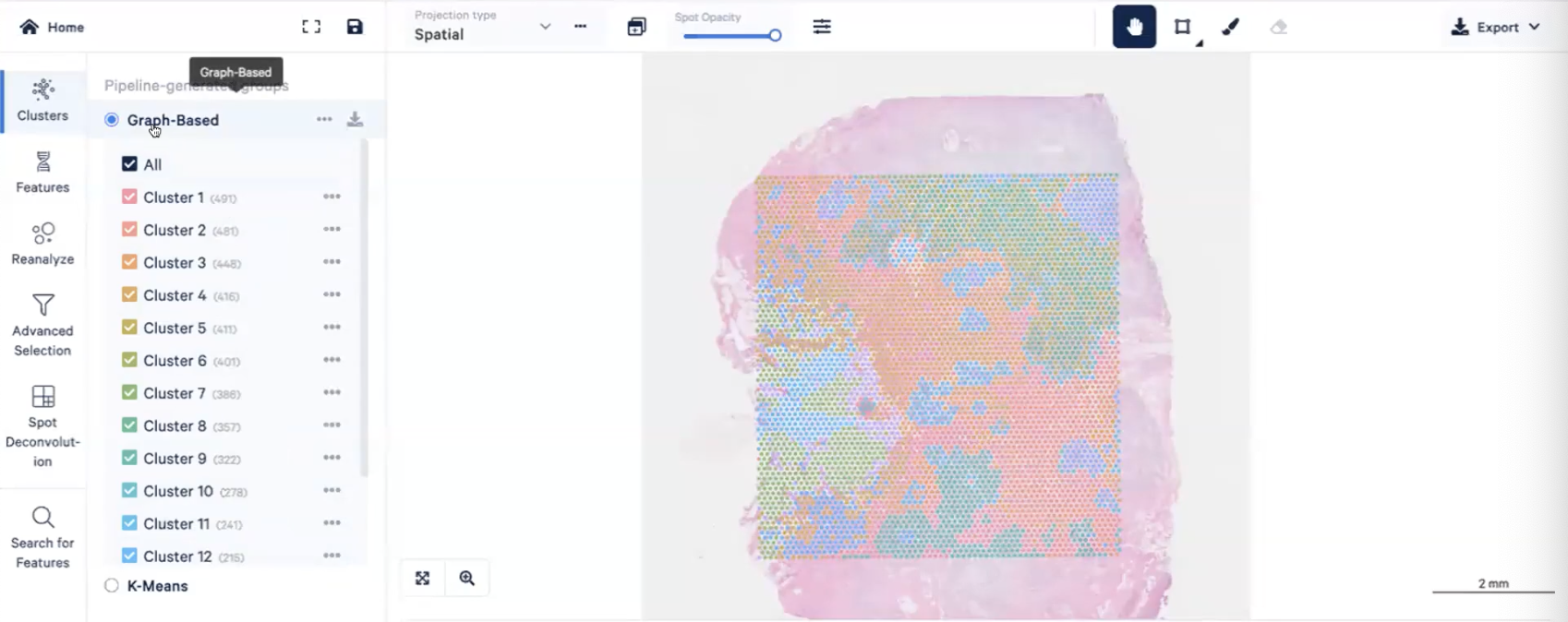
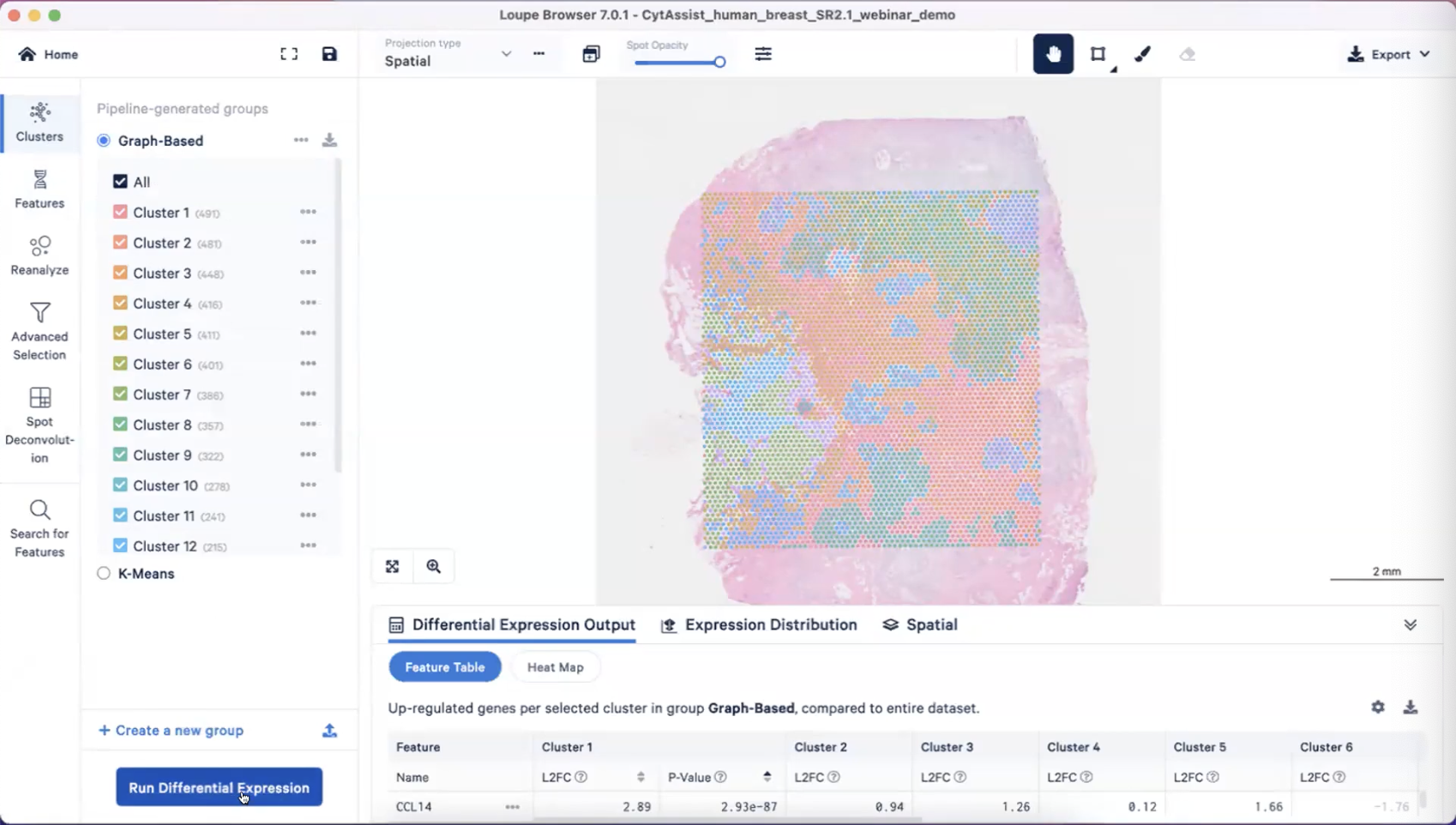
- 左侧的功能栏给我们提供了不同的功能,cluster功能栏展示了不同的聚类结果,Loupe Browser提供两种聚类算法,分别是Graph-Based和K-Means,其中K是我们期待的聚类数目(K值可以从2-10)
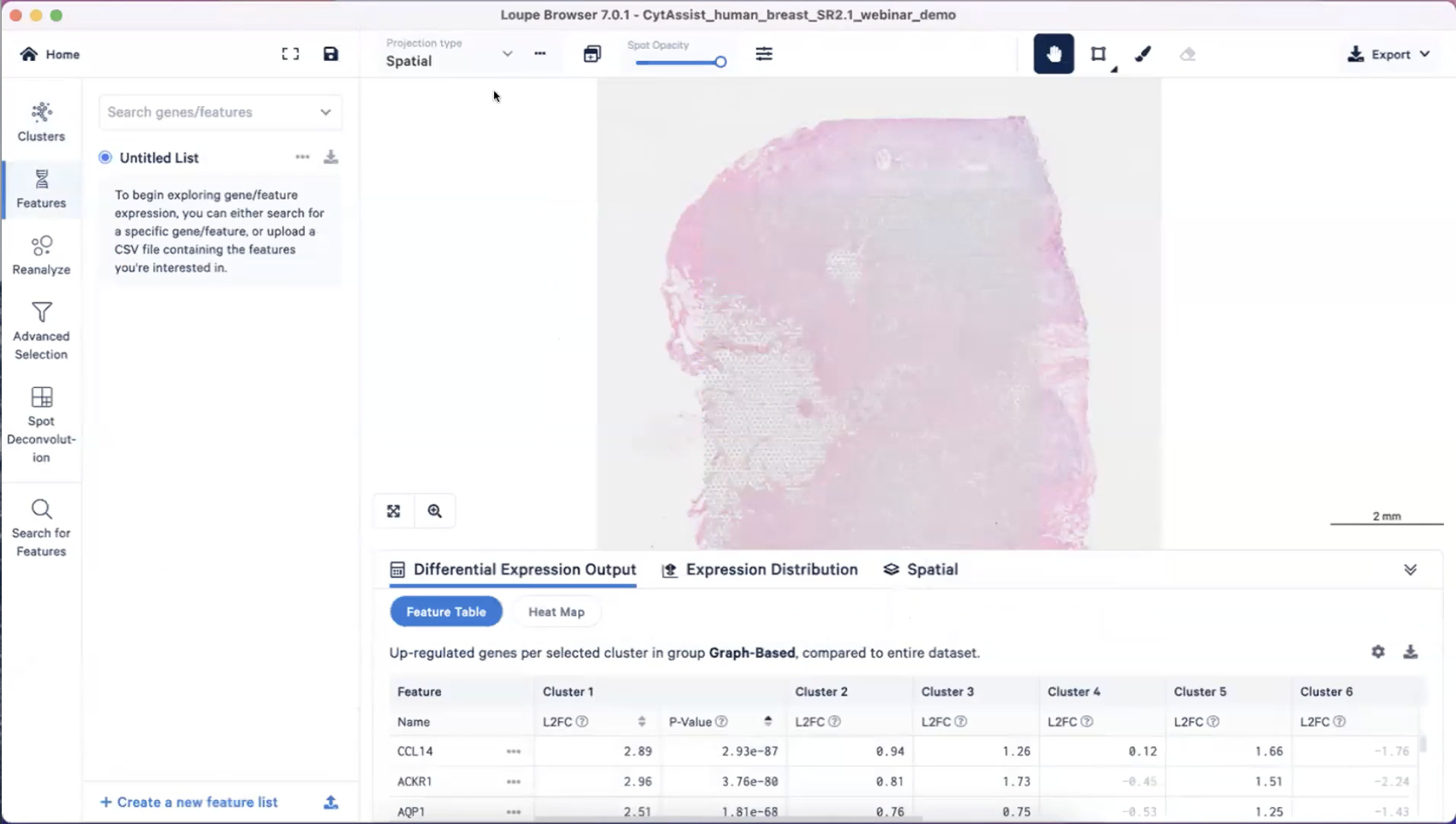
- cluster栏下方的”Run Differential Expression”按钮可以供我们进行差异表达基因分析,计算出每个cluster高表达的基因,这些基因可能是marker gene
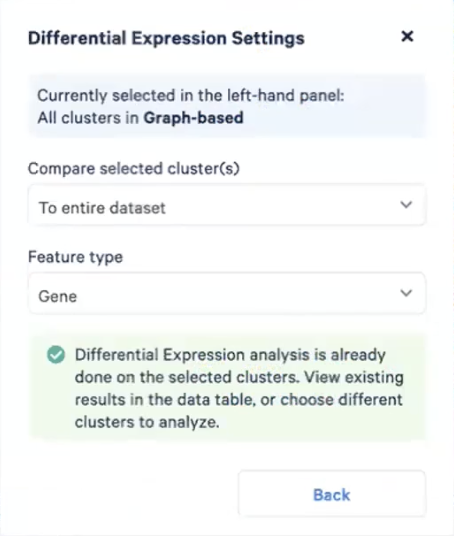
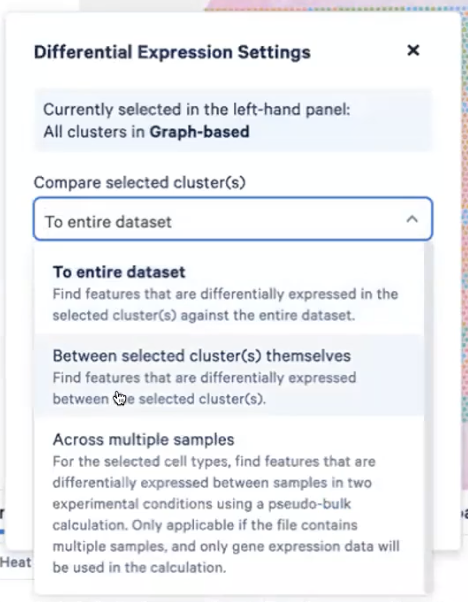
- 不同的计算方法
- to entire dataset:每个cluster和其他cluster进行比较,去计算高表达的基因
- between selected cluster(s) themselves:我们自己选一些cluster,然后在这些cluster之间比较
- across multiple samples:在多样本间进行比较,探究样本间而不是样本内的差异因素(比如用药、耐药、治疗后)对细胞产生的影响,这个功能是通过pseudo-bulk DEG算法去实现的。
feature功能栏
- 接下来是feature功能栏,在这个部分我们可以探索感兴趣的基因在整张组织切片上的表达情况,这张切片来自HER2阳性的乳腺癌标本,所以我们可以探究HER2(也叫ERBB2)在这张切片上的表达情况,HER2高表达的区域在这张切片上被染成深红色,我们去放大这些区域后,会发现高表达HER2的区域确实存在着很多肿瘤细胞
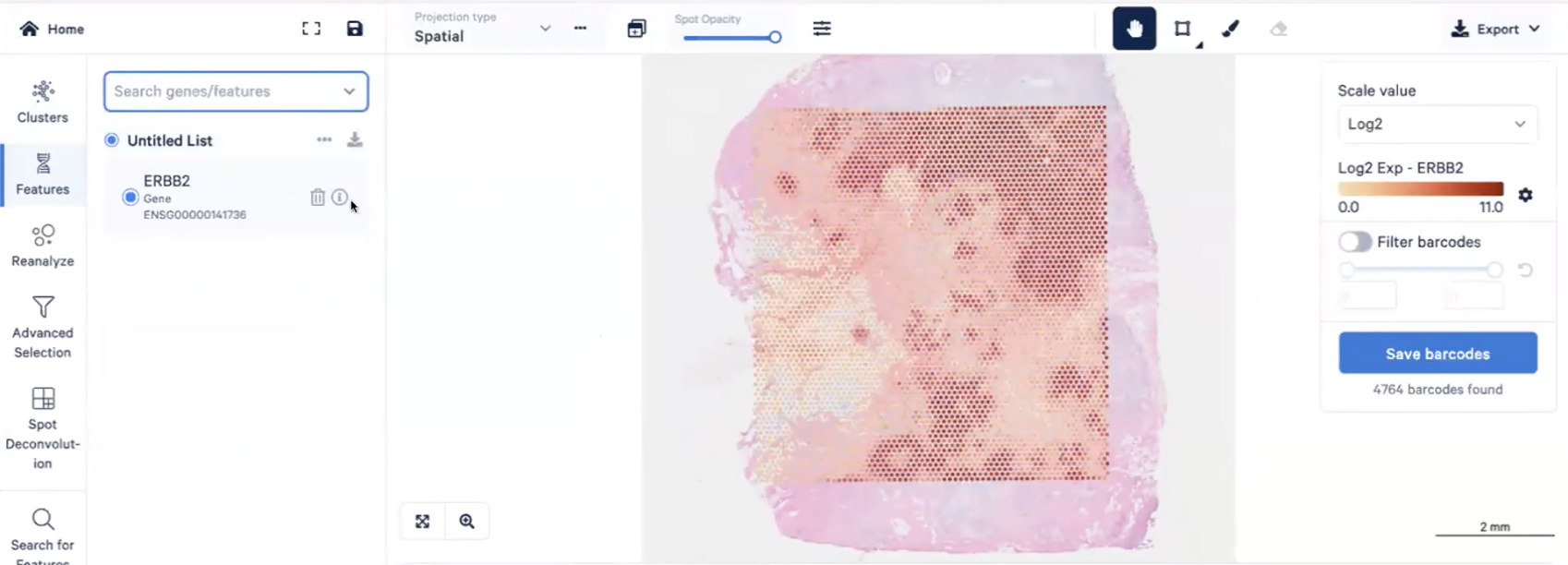
Reanalyze功能
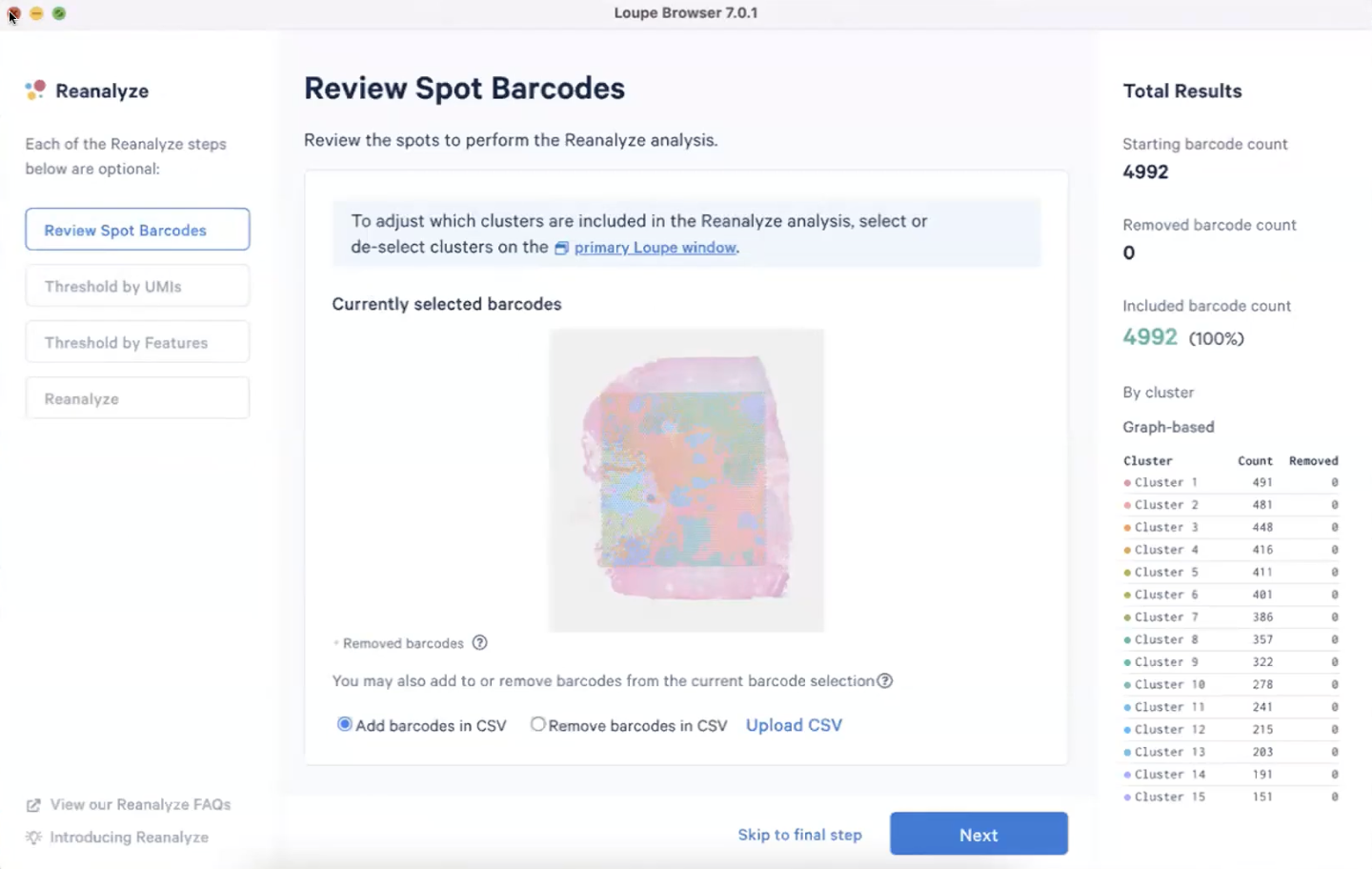
- 第三个功能栏Reanalyze可以让我们对Visium数据进行一定的QC,具体见下
Advanced Selection功能
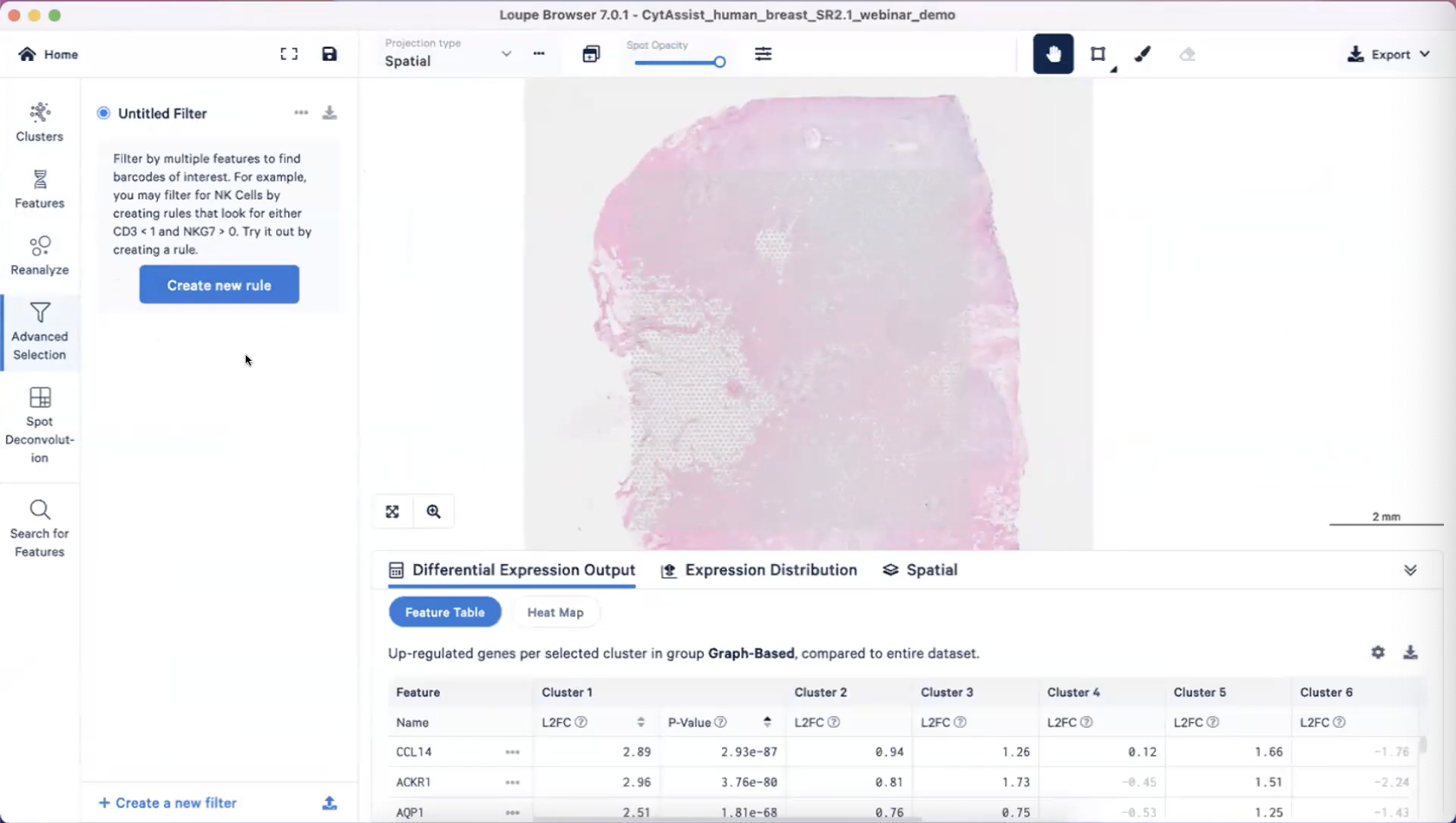
- 我们可以自定义一些规则,从而更高效地选择我们感兴趣的细胞,具体见下
Spot Deconvolution功能
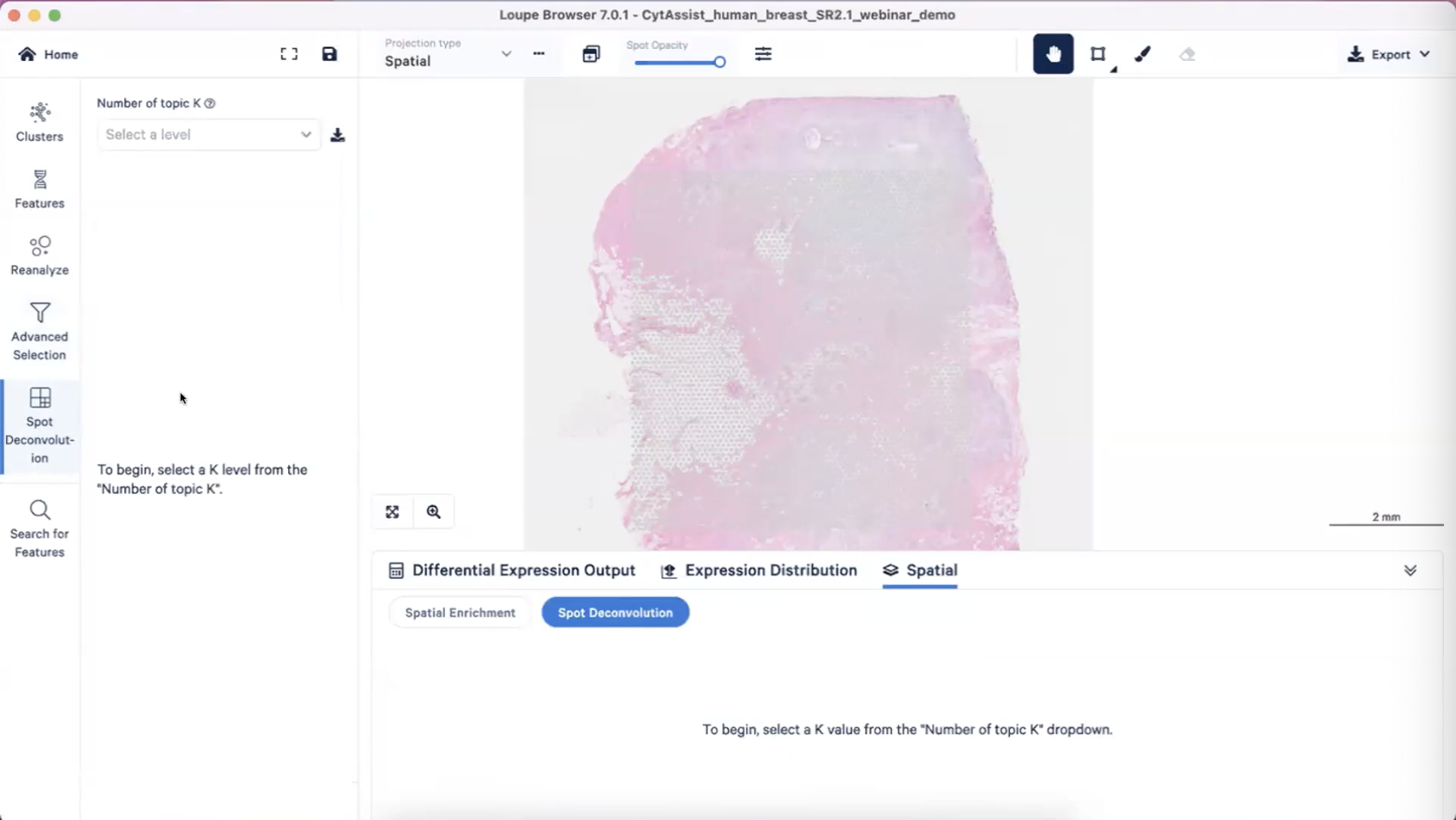
- 在这个部分,10x引入了LDA modeling的算法来进行reference-free spot deconvolution,如果没有单细胞数据,算法可以自动帮我们的Visium数据进行spot deconvolution,如果打开的数据没有Spot Deconvolution功能界面,可以选择把fastq文件在2.1版本以上的Space Ranger中进行
- LDA modeling算法的缺点是这个算法不能识别出样本中的cell type的数量,所以需要输入cell type的数量,这个值被称为K值(和前面的K-means相同),这里提供的K值范围是2-17,17是Graph-Based产生的cluster值+2
- 在实际工作时,需要我们去探索输入的K值产生的deconvolution结果,在这个示例中,如果我们指定K值为4,那我们可以得到一张类似于下图的结果,下图中的热图上的每个点的值在0-1之间,越接近1说明我们指定的细胞类型在这个spot中的占比越高,越接近100%
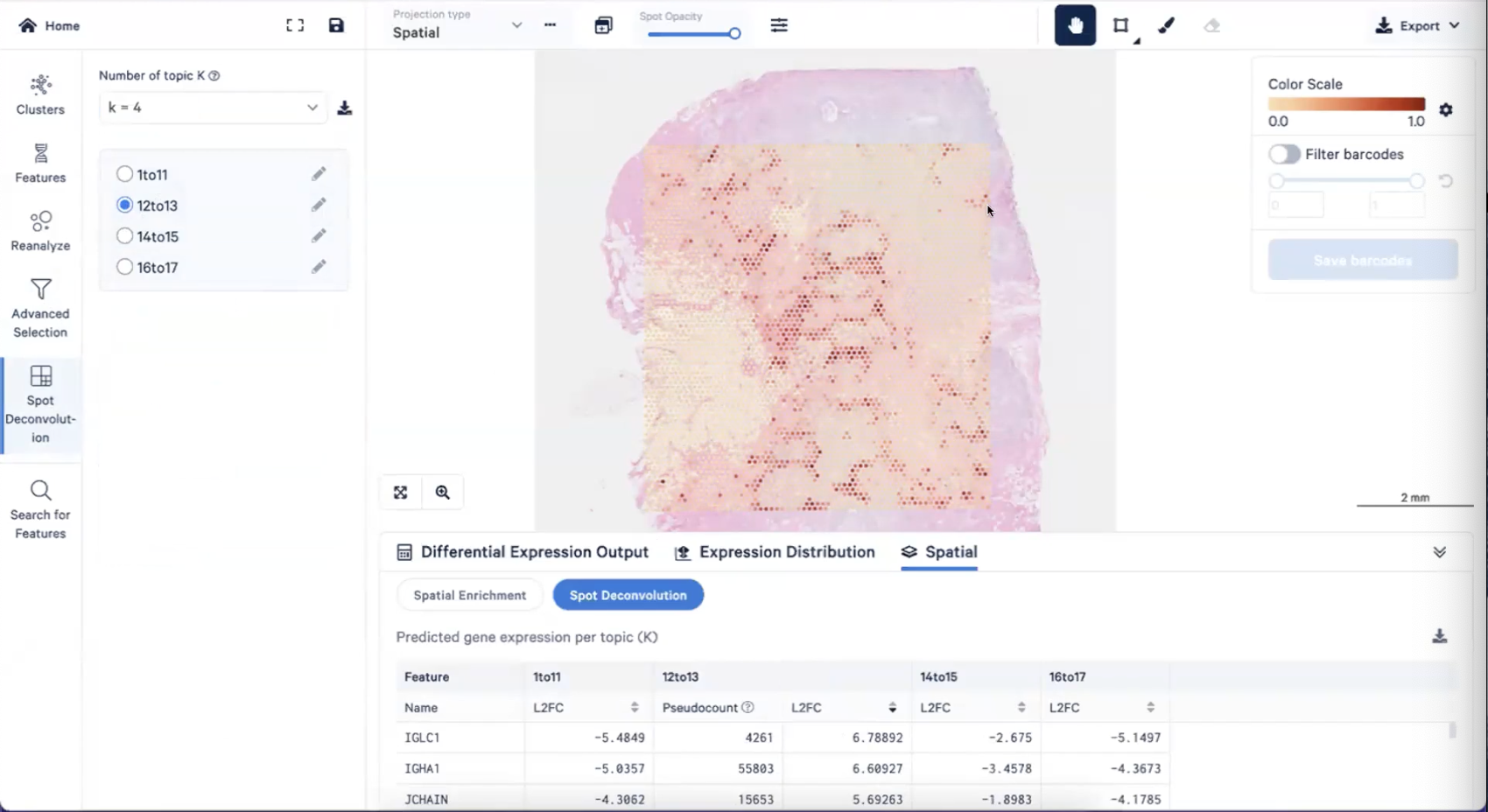
- 如果我们想要了解我们所选中的”12to13”组细胞的具体细胞类型,我们可以关注界面下方功能栏中的Spatial-Spot Deconvolution列表,可以看到我们关注的”12to13”组细胞有Pseudocount和L2FC两个参数,一些与这个组细胞高度相关的基因可以被列举在表格中,我们发现第二组高度相关的基因都是免疫相关,所以提示我们这个组的细胞很有可能与免疫相关;通过高表达的基因(也就是所谓的marker gene)的注释,我们可以发现每个spot中每种类型的细胞的组成
Differential Expression Output
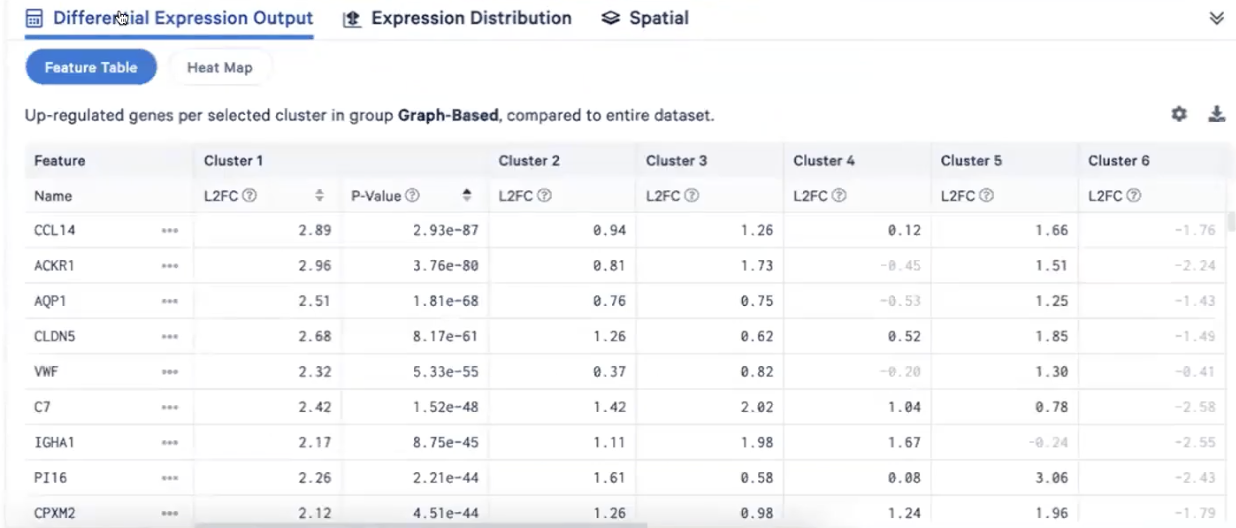
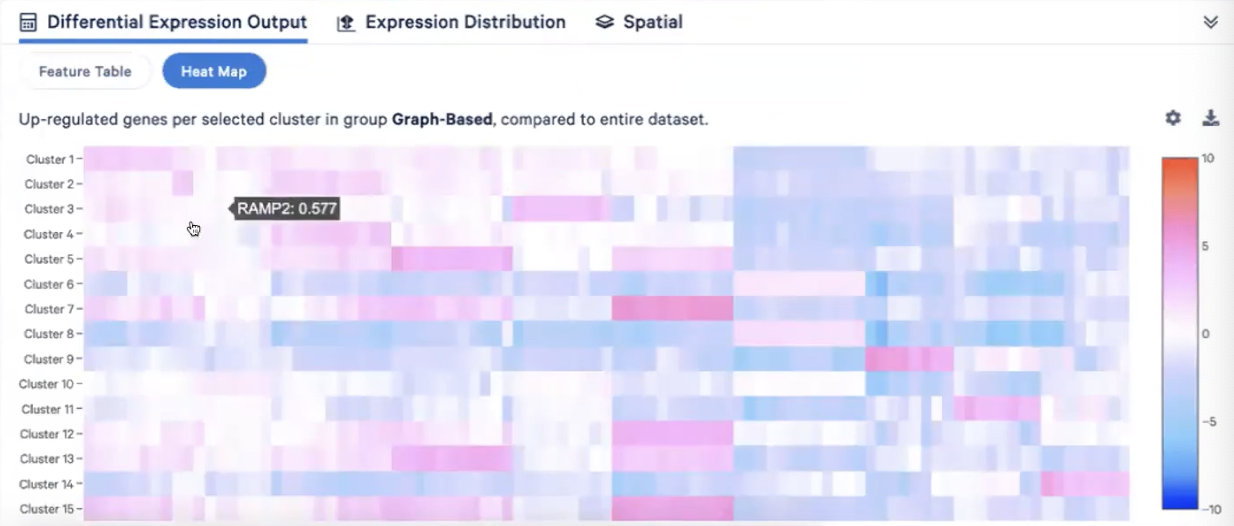
- 展示的是每个cluster的marker gene,包括L2FC和FDR值(这里的p值是经过检验的),这个部分的内容也可以用热图的形式去呈现,每个横列是cluster,纵列是marker gene,颜色代表的是基因表达的情况(z-score值)
Expression Output功能
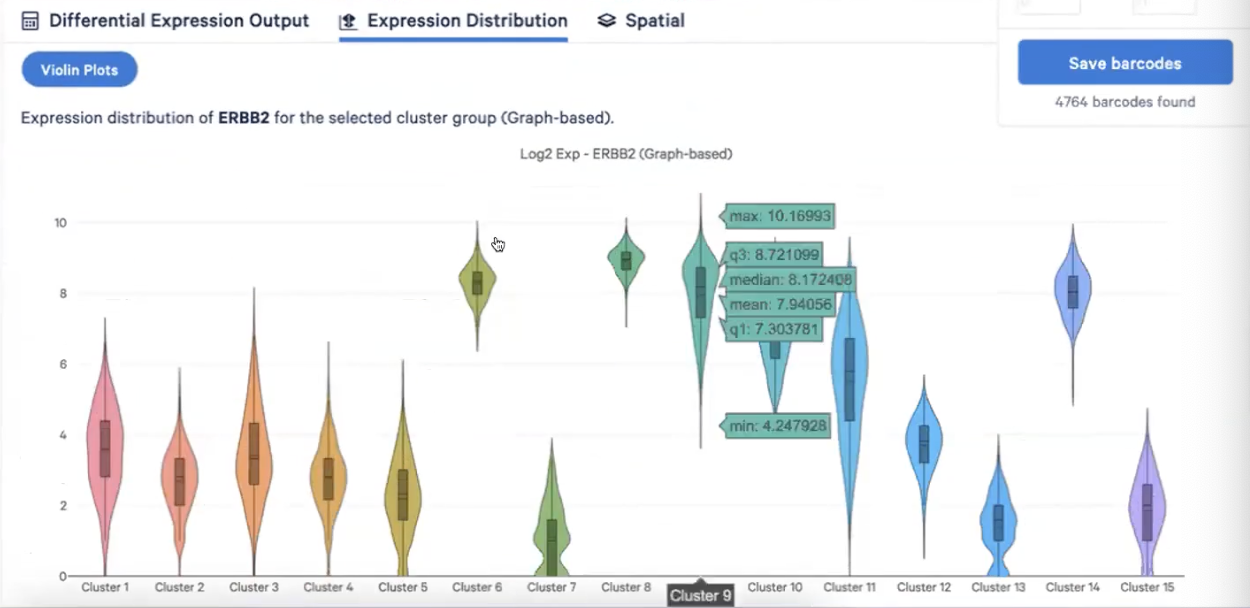
- 我们可以在这个部分观察某个基因(比如我们指定的ERBB2)在每个cluster中的表达情况
Spatial功能
- Spatial的功能一共有两部份,分别是Spatial Enrichment和Spot Deconvolution,后者的介绍见前,这部分主要讨论Spatial Enrichment的内容
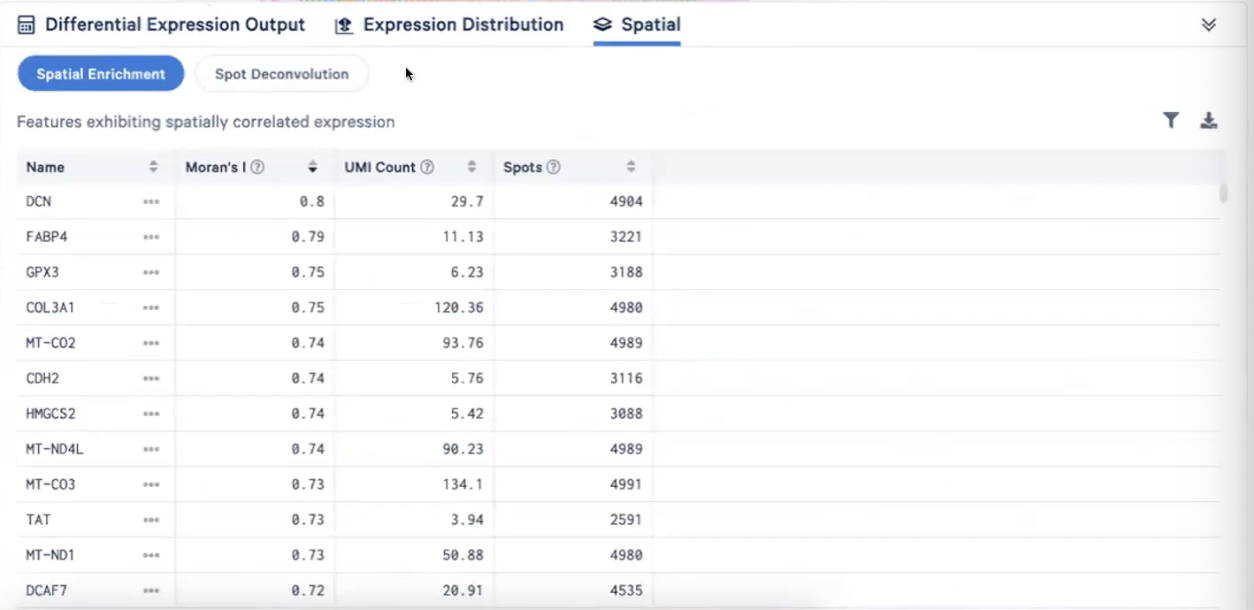
- Spatial Enrichment主要展示的是Moran’s I index,这个值是cluster-free auto-correlation的统计学算法,在正常情况下这个值在-1到1之间变化,越接近-1就说明这个基因的分布是完美离散的,越接近0说明这个基因的分布是随机的,越接近1就说明这个基因的分布是集中在某个区域的
- 在Loupe Browser中展示的Moran’s I index的范围是0-1,越接近0说明这个基因在空间上的表达越随机,越接近1就说明这个基因的分布是集中在某个区域的
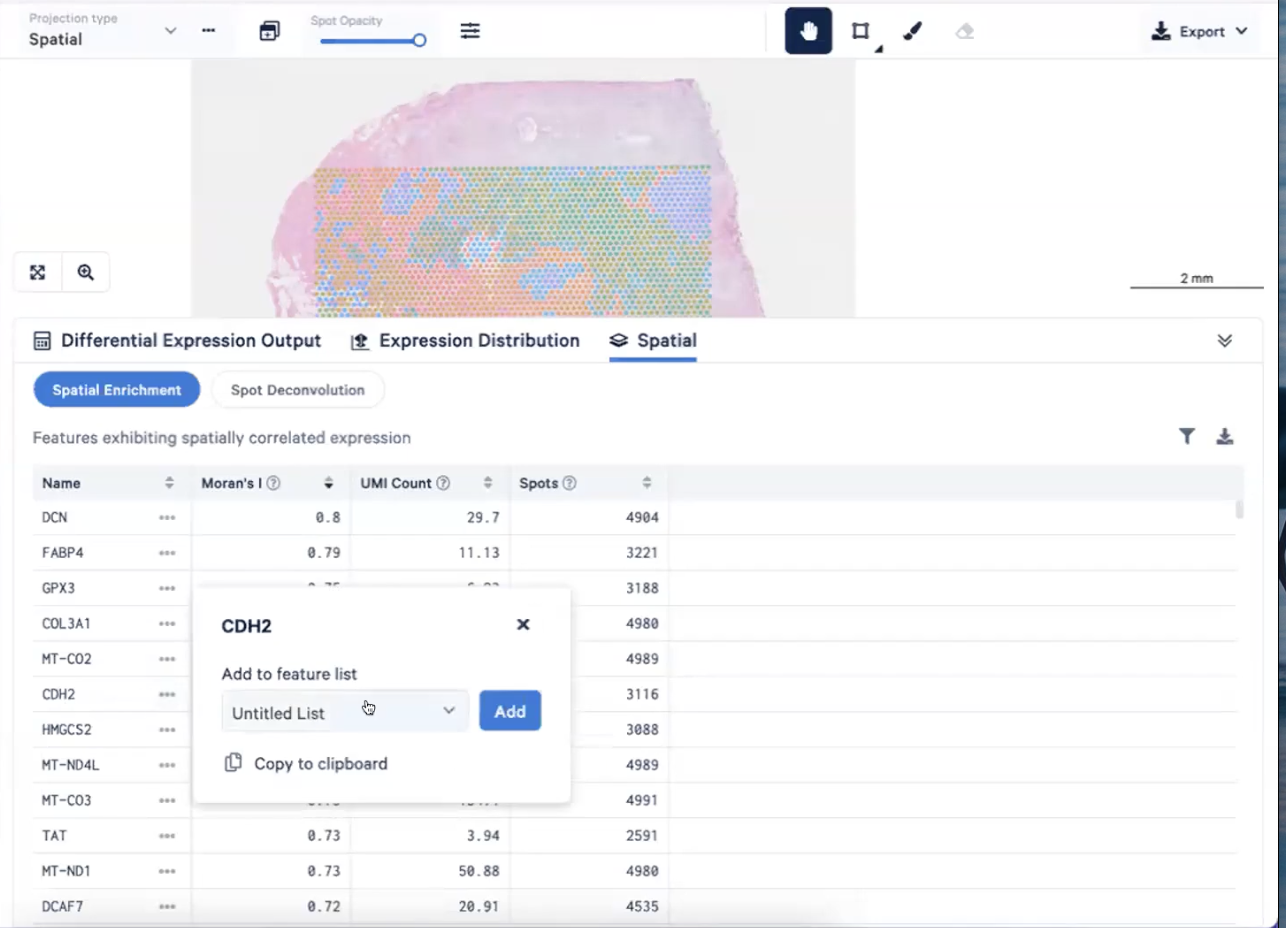
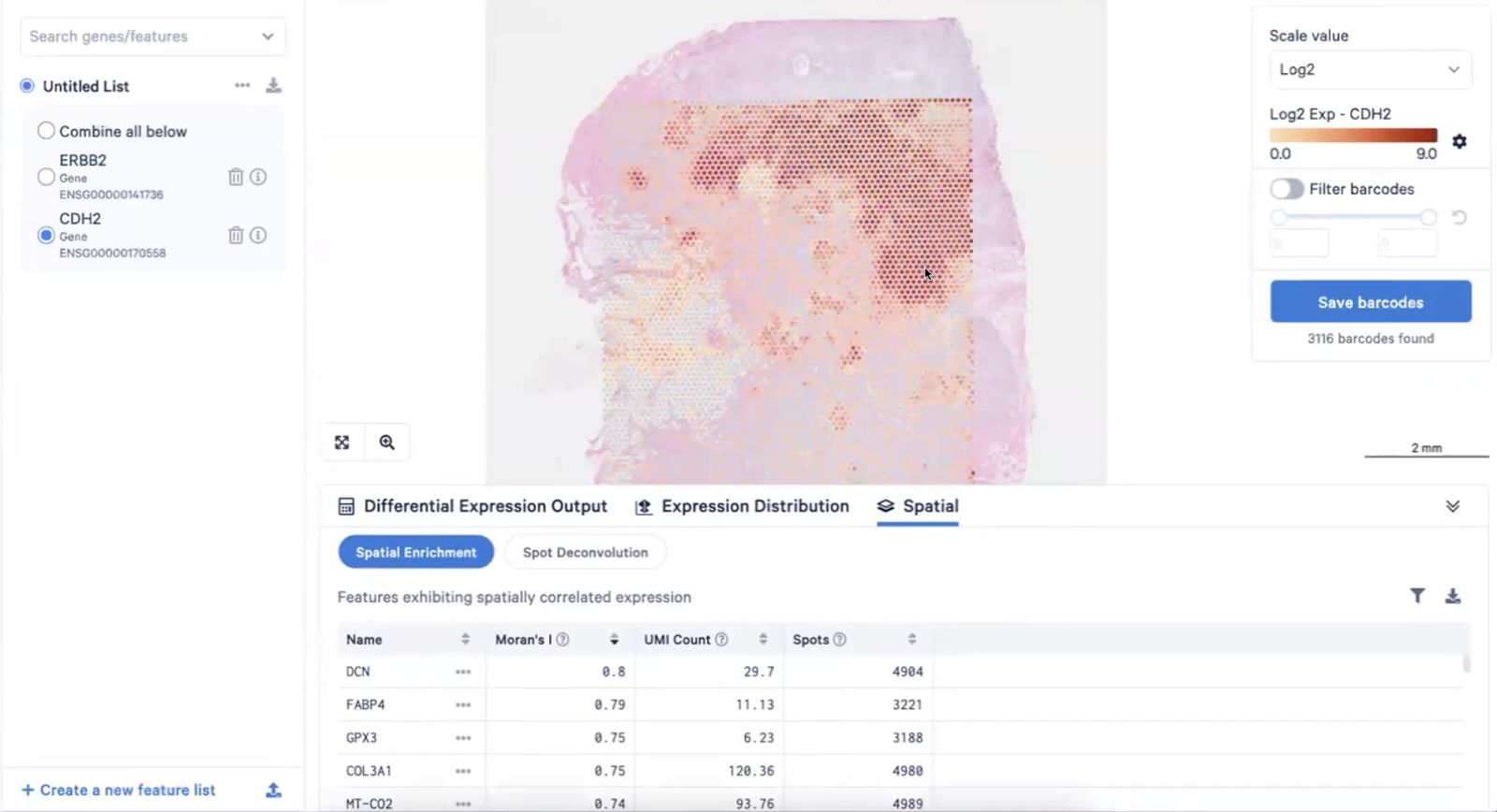
- 例如上图中展示的CDH2,又称N-cadherin,大量参与EMT过程,可以在恶性肿瘤中出现,可以看到CDH2的值高达0.74,提示这个基因在很大程度上局限于这个切片的某个区域,而当我们近距离观察这个区域,发现这个区域有很多invasive cancer

- 大家看到高比例的线粒体基因不用害怕,因为肿瘤组织会需要有高水平的线粒体进行呼吸作用,高比例的线粒体基因出现在肿瘤细胞中是生物学上可以解释的现象。
使用Loupe Browser进行数据分析
简单的QC
- 使用Reanalyze功能,下图的右边会告诉我们有多少个spot被去除和选中
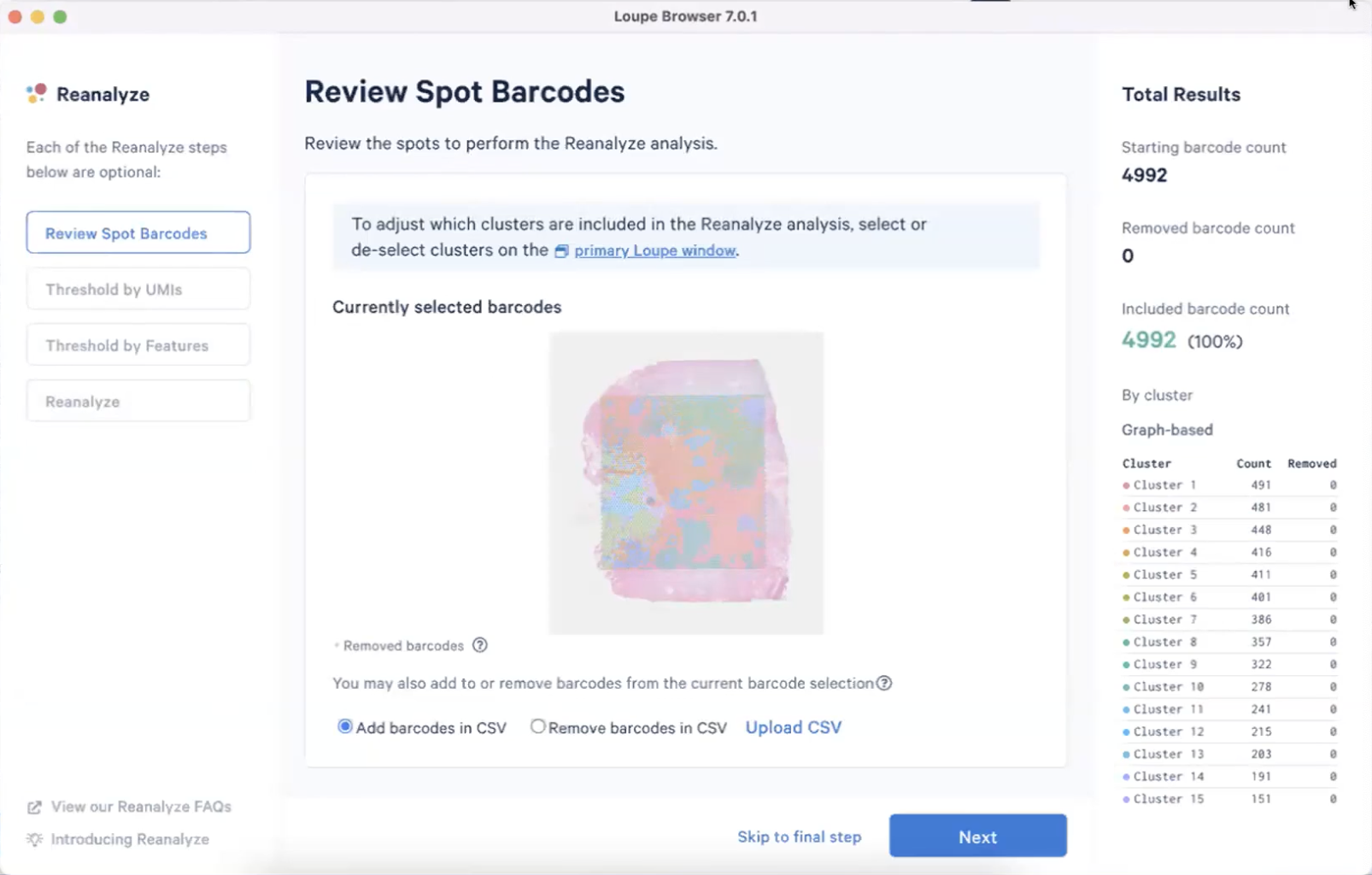
- 在Visium数据中,我们主要根据每个spot的UMI数和基因数量进行QC,首先对于UMI数量进行QC,在这一步我们推荐对UMI的数量进行Log2转化,如下图所示
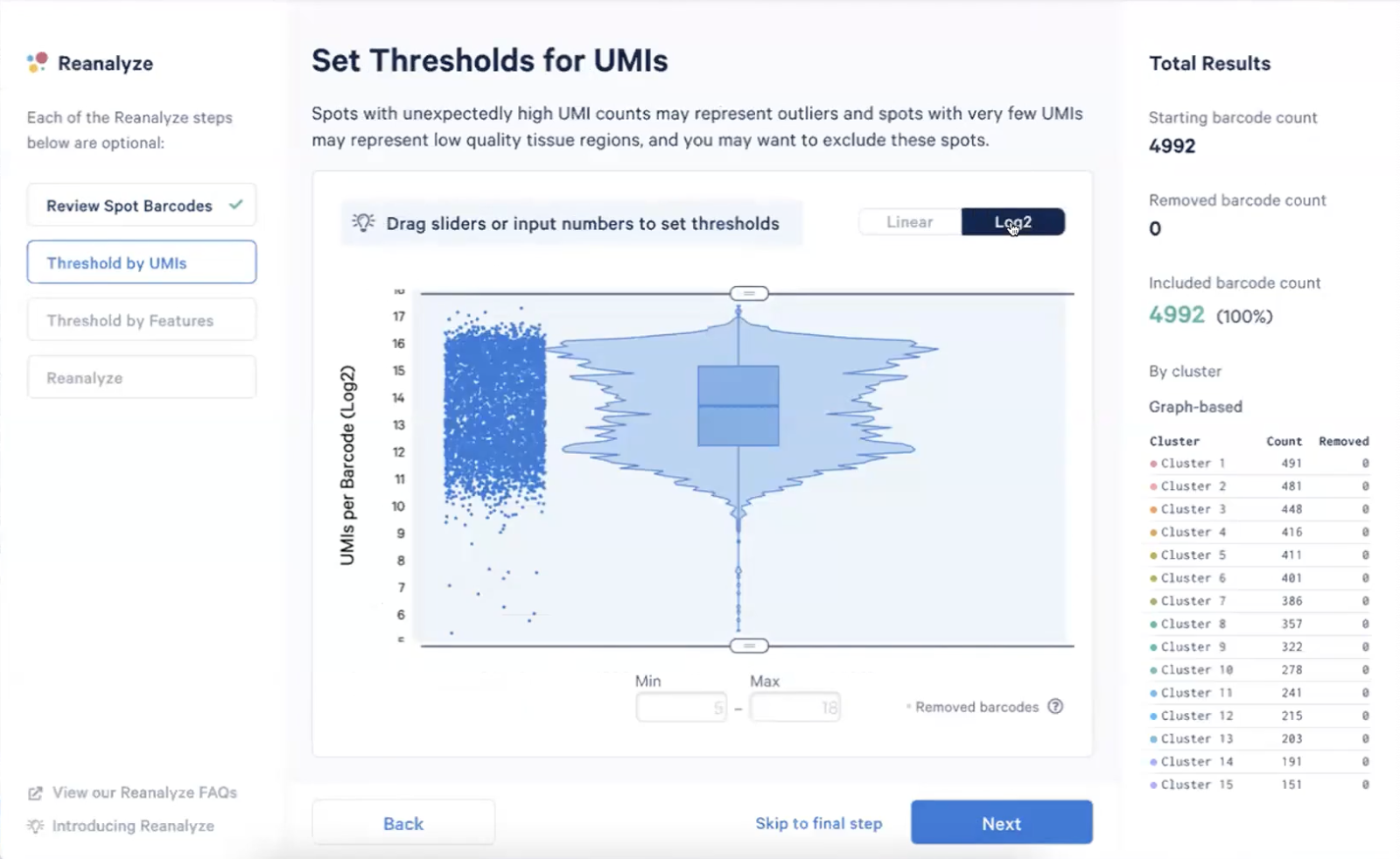
- 我们可以拉动来调整下限值,对于那些UMI数量非常少的spot,可能提示这些区域覆盖了一些低质量的组织,我们也可以手动选择下限值
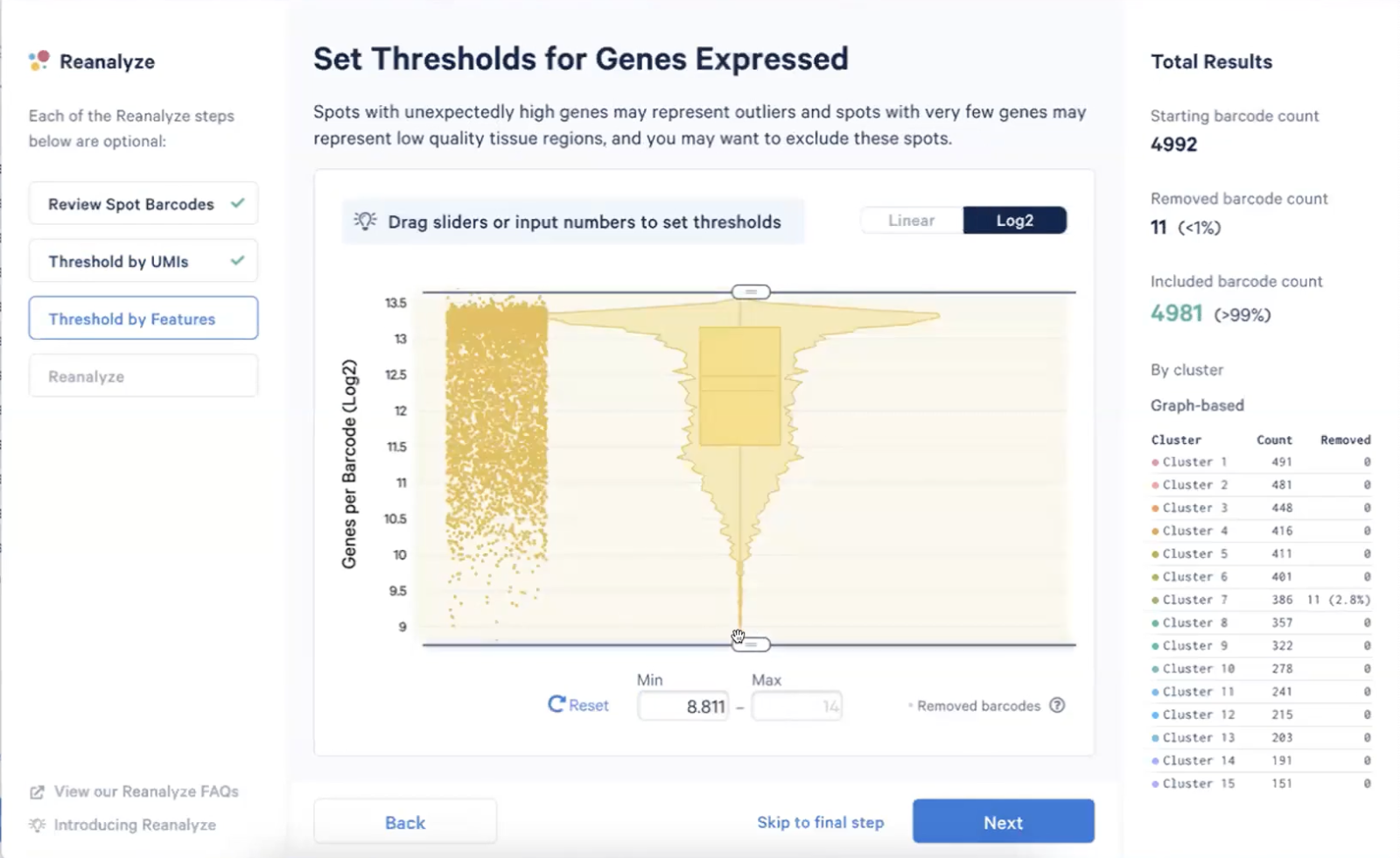
- 接着,我们会对每个spot的基因总数是QC,我们也推荐对纵轴进行Log2转化,结果如下图所示,我们在这一步也要去除基因表达数目非常低的spot,我们可以进行手动拖拽或输入下限值来进行这一步的QC
- 在两步QC之后,我们需要对样本重新计算t-SNE和UMAP的结果,并且重新命名为一个组别,然后进行recluster,如下图所示
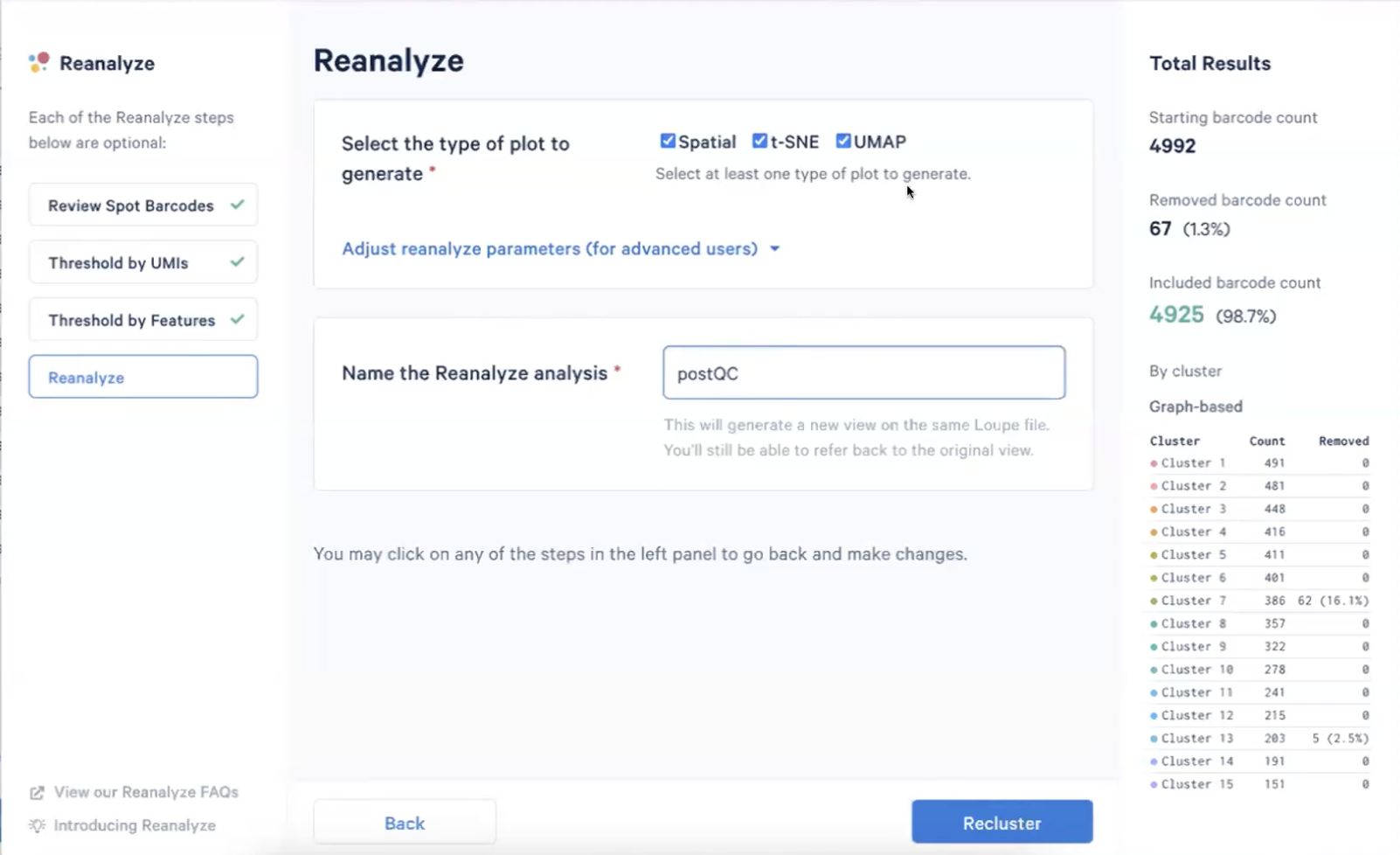
- 这两步QC的目的是移除那些UMI和基因数特别低的cluster,去除这些可能低质量的区域有助于我们进行进一步的数据分析,减少背景的干扰,拿到更好的clustering和dimentional reduction的结果。
- 成功之后,可以看到在Clusters栏中多了一个postQC组,如下图所示
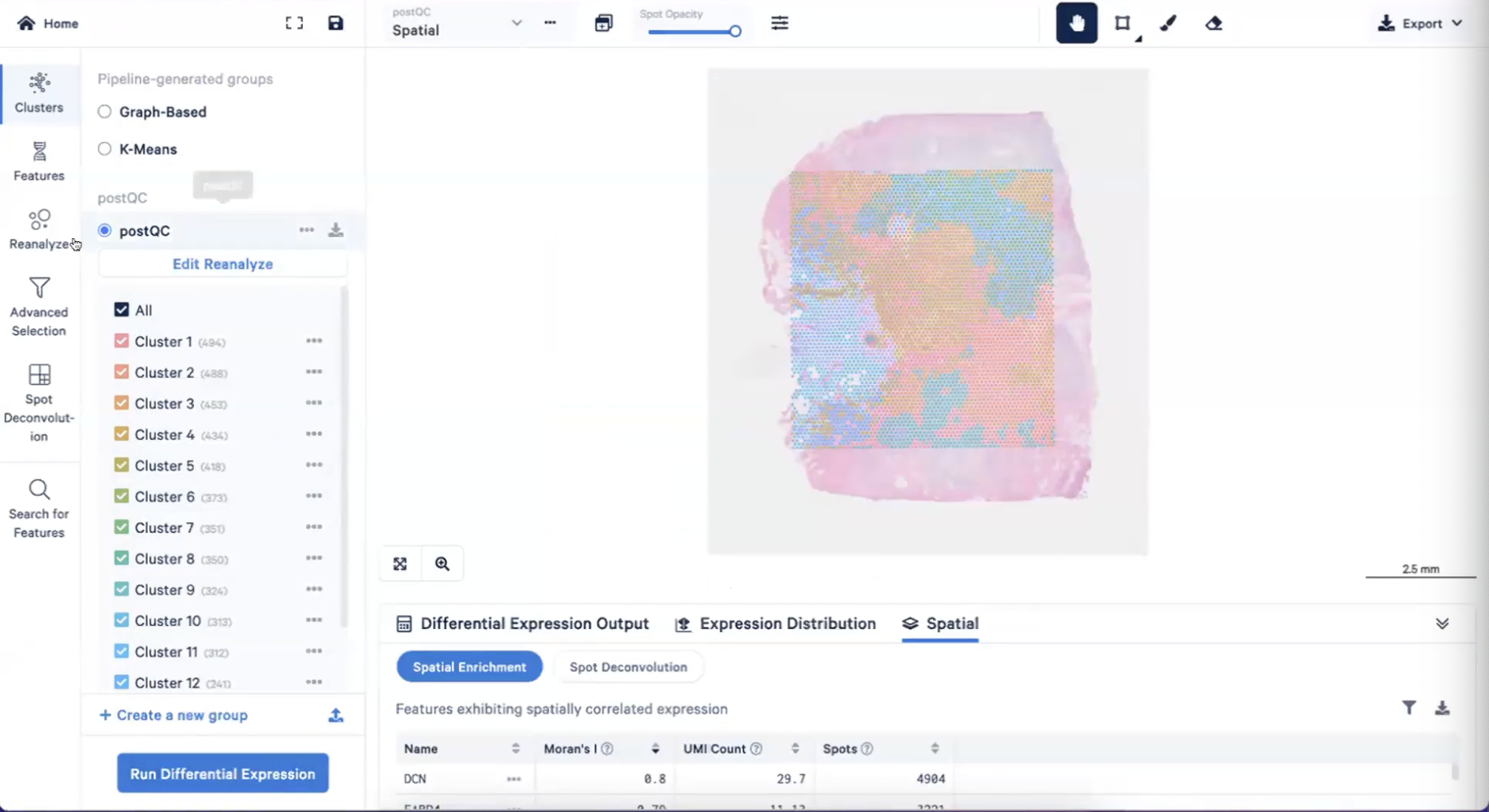
- Loupe Browser也支持展示post QC的Spatial/t-SNE/UMAP图
- 在QC后,需要重新计算DEGs从而获得新的cluster的marker gene,了解每个cluster的marker gene
spot注释
- 我们首先尝试去注释肿瘤细胞,样本来源是HER2(+)的乳腺癌,因此可以用HER2去注释肿瘤细胞
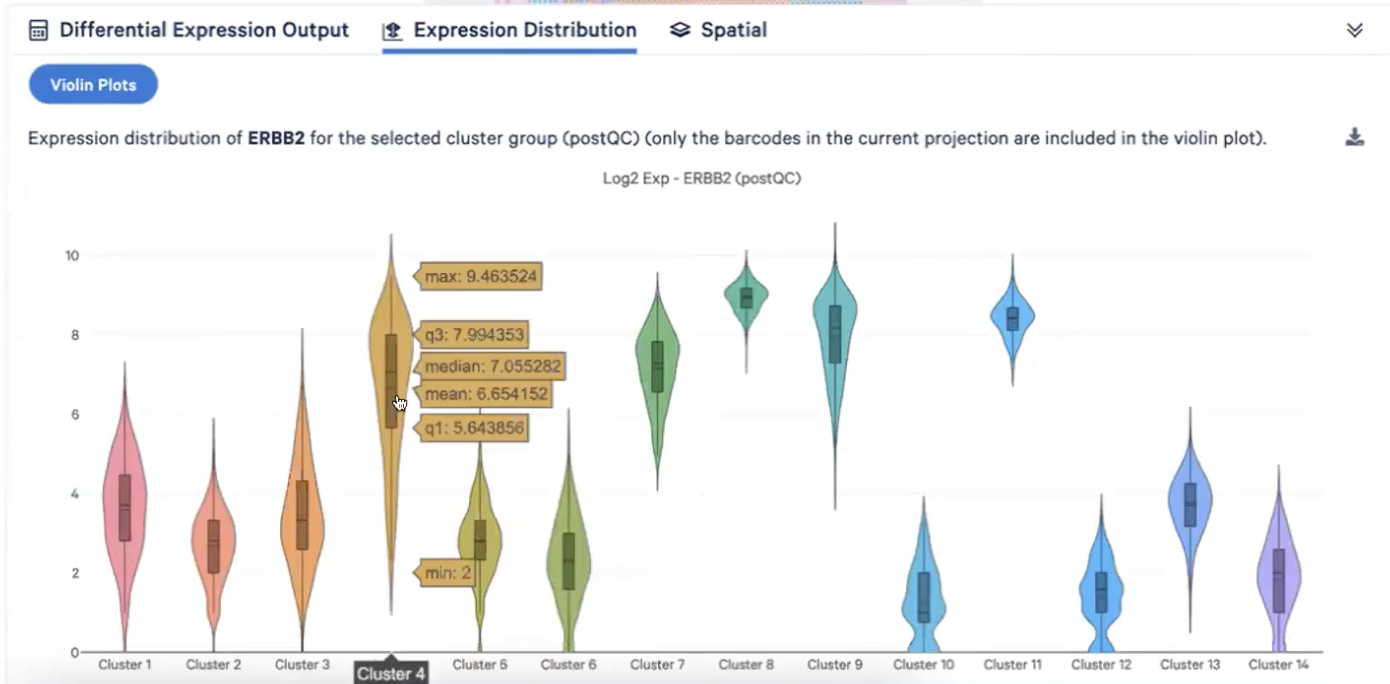
- 在feature工具栏中可以选择HER2,然后观察下方工具栏的Expression Distribution,如下图所示可以发现HER2高表达在cluster4、7、8、9、11中,我们可以判断这些高表达区域的HER2的cluster所覆盖的区域确实是肿瘤细胞所在的区域
- 补充知识:如果我们想判断上图的cluster所对应的肿瘤的进展情况
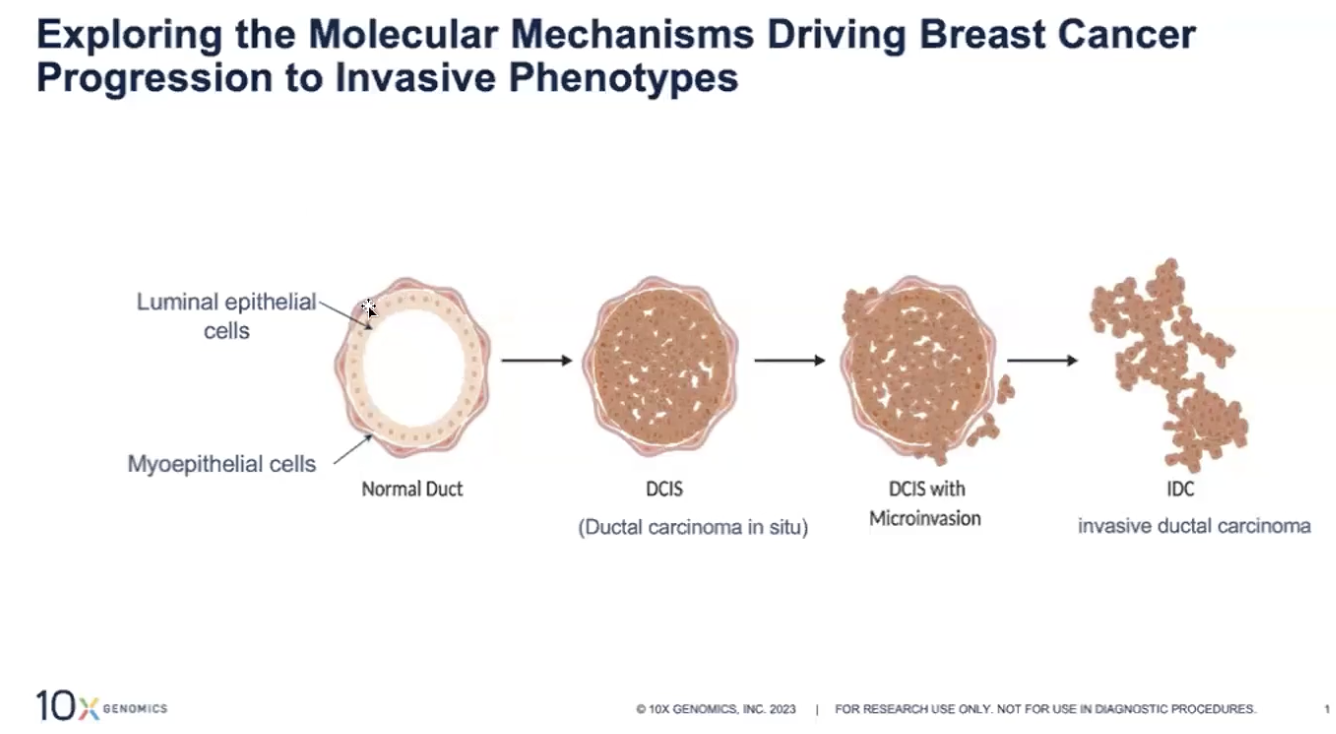
- 最左边的图是人类乳腺正常的导管,最内层是管腔上皮细胞,其外层围绕着肌上皮细胞和更内层的基底膜,通常我们的乳腺癌是从导管内皮细胞发生的,导管内皮细胞在管腔内不断增殖但尚未突破管腔,在肌上皮尚完好的状态称为导管原位癌,属于较早期的肿瘤
- 如果肌上皮的结构被突破,肿瘤向周边的正常组织进行浸润,这种时候我们就可以认为肿瘤细胞逐渐恶化,形成了侵袭性的导管腺癌
- 回到我们的样本上来,我们可以学习到肌上皮在肿瘤组织中的存在可能提示肿瘤处于早期,所以我们可以观察样本肿瘤区域中肌上皮的marker,也就是KRT17去探索对应区域的肿瘤细胞的特点
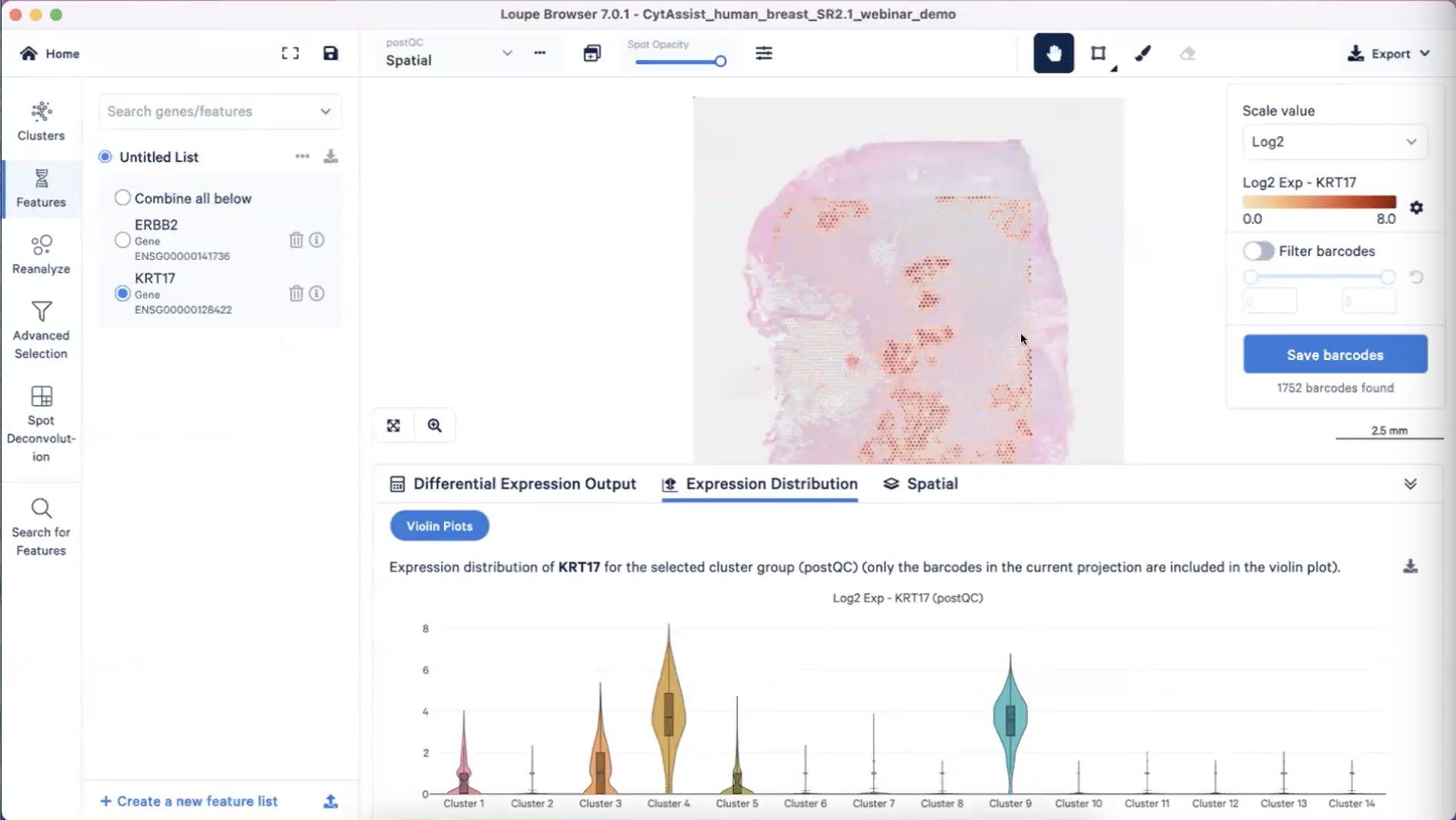
- 上图中,我们可以看到KRT17出现在了cluster4和9,且这些区域的KRT17表达出现了明显的“边界”,而HER2表达出现在了cluster4、7、8、9、11,所以cluster4和9可能覆盖了导管原位癌所在的区域
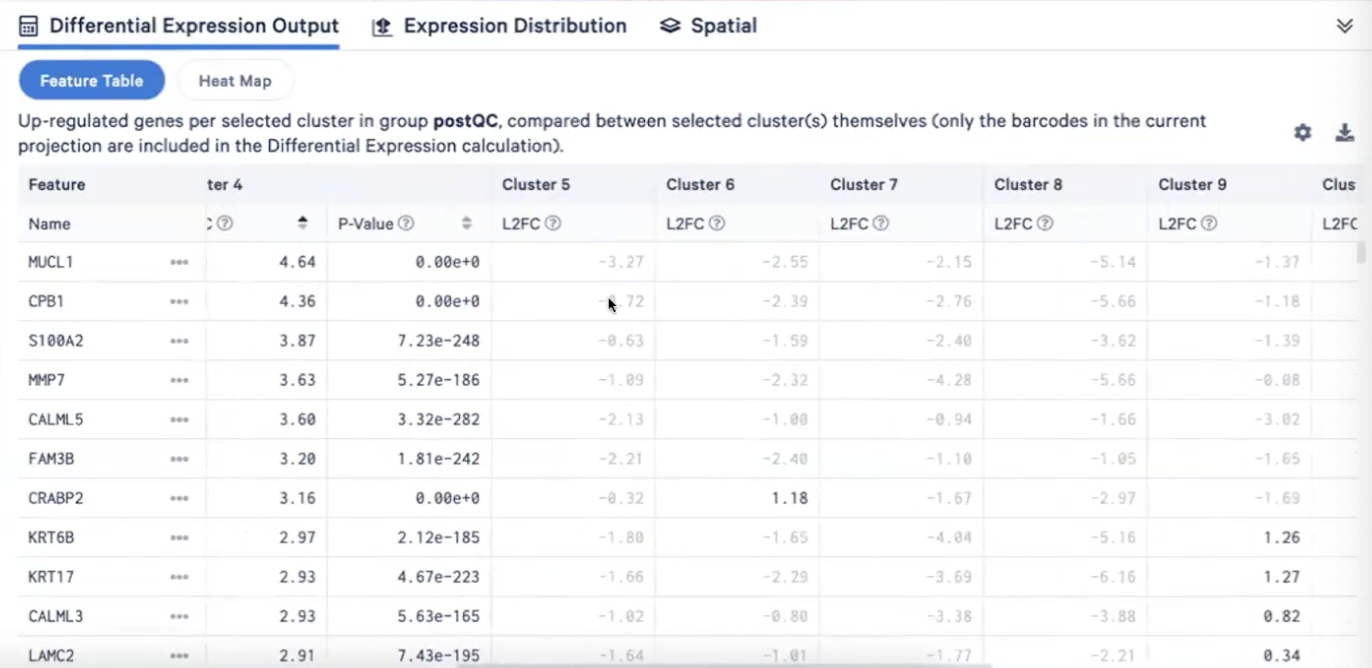
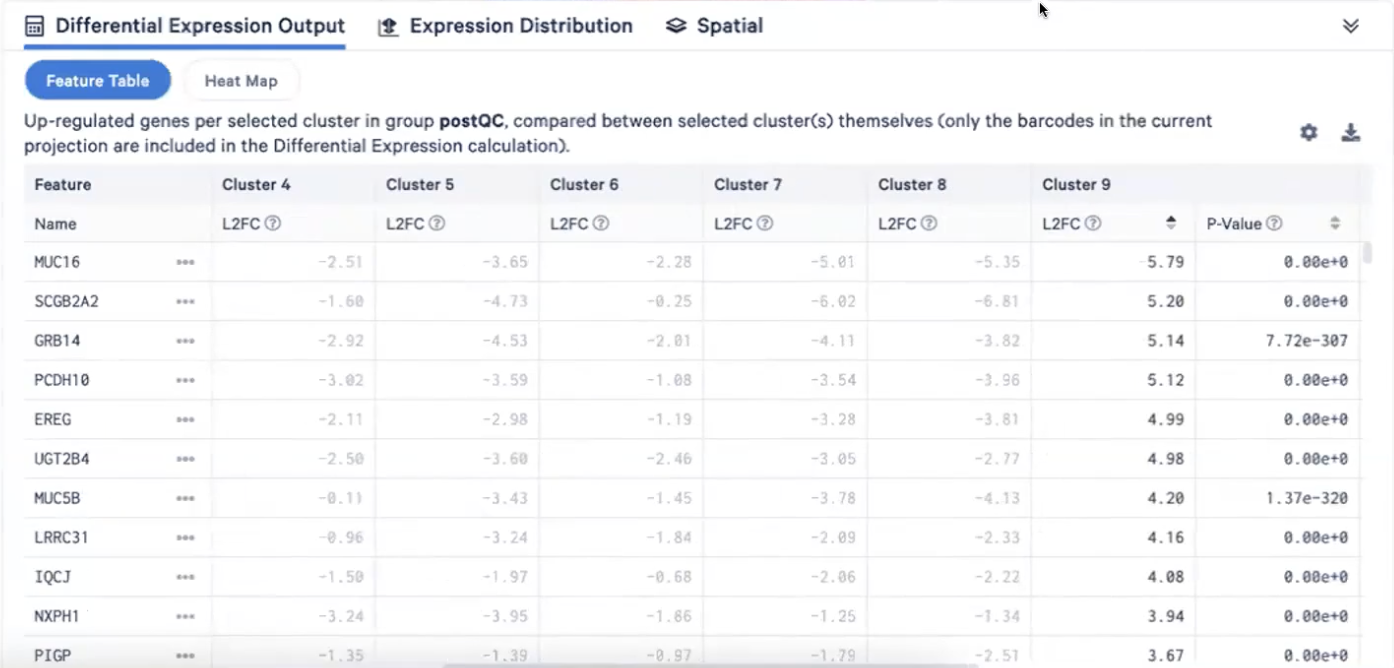
- 我们可以进一步探索cluster4和9的差异表达基因,比如说cluster4高表达CPB1,而这个基因可能与乳腺癌的某种预后相关(下图1);对于cluster9,我们发现它高表达SCGB2A2这个基因,而这个基因的表达可能跟骨转移相关
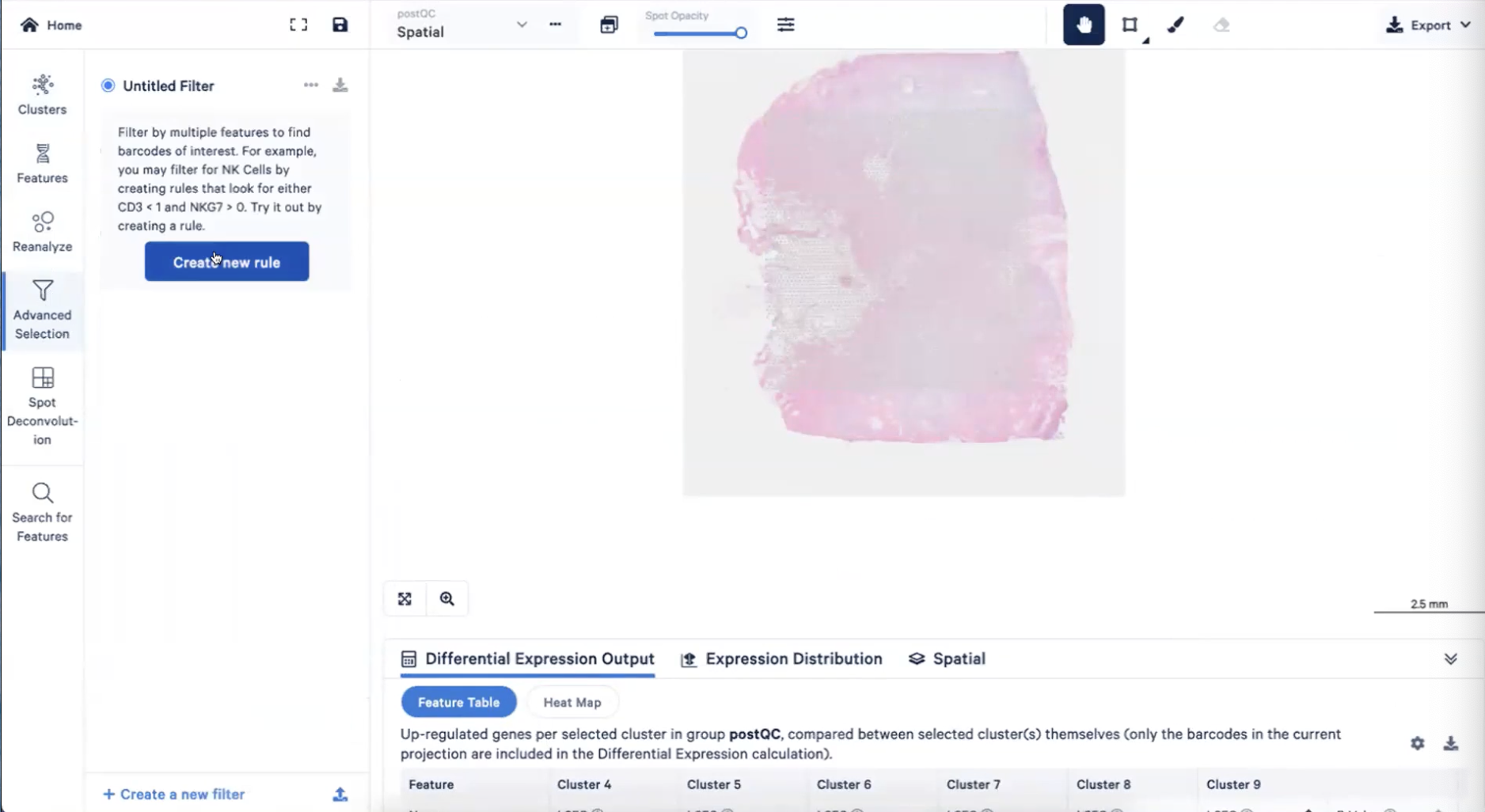
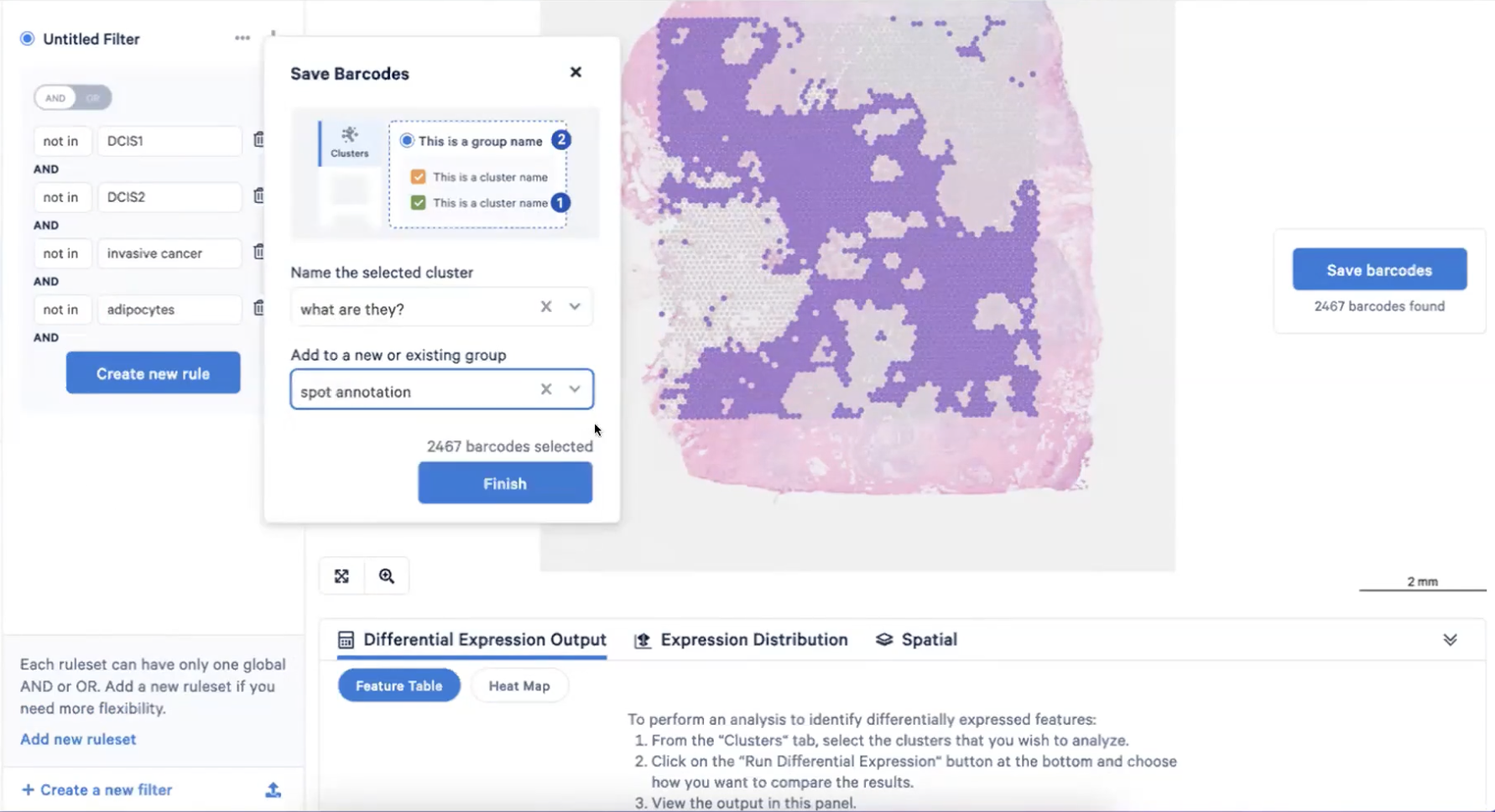
- 总结来说,这两群细胞的表达差异是非常大的,因此我们进入到细胞注释步骤的时候,就可以把这些基因给分开来,具体来说我们可以进入到advanced selection界面,选择”Create new rule”-Cluster 4
- 然后我们选择in cluster 4-右侧的Save barcodes-把这个cluster命名为导管原位癌1型(DCIS1),并创建一个新的group,命名为spot annotation,如下图所示
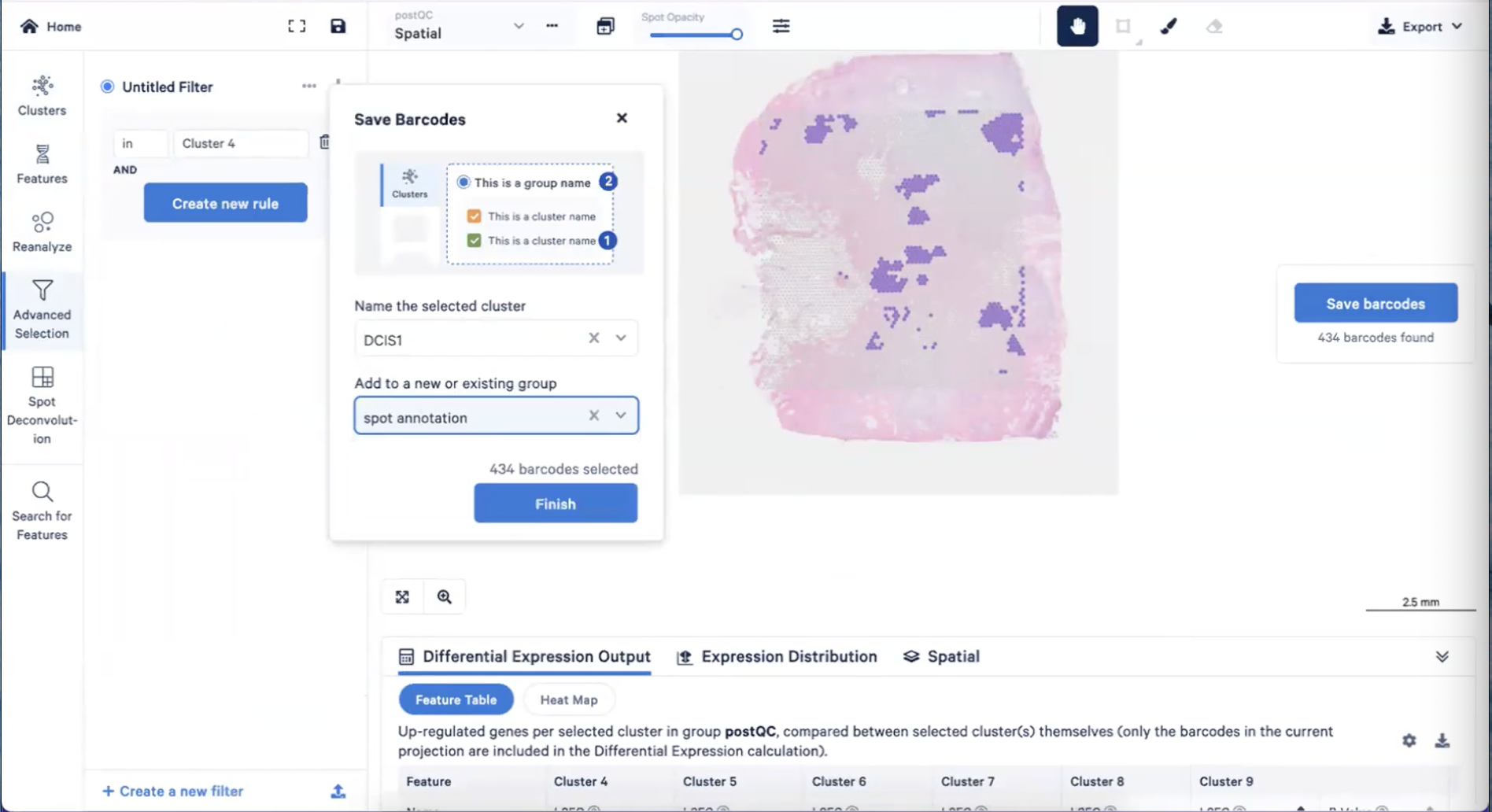
- 相对的,我们可以把cluster9注释为导管原位癌2型
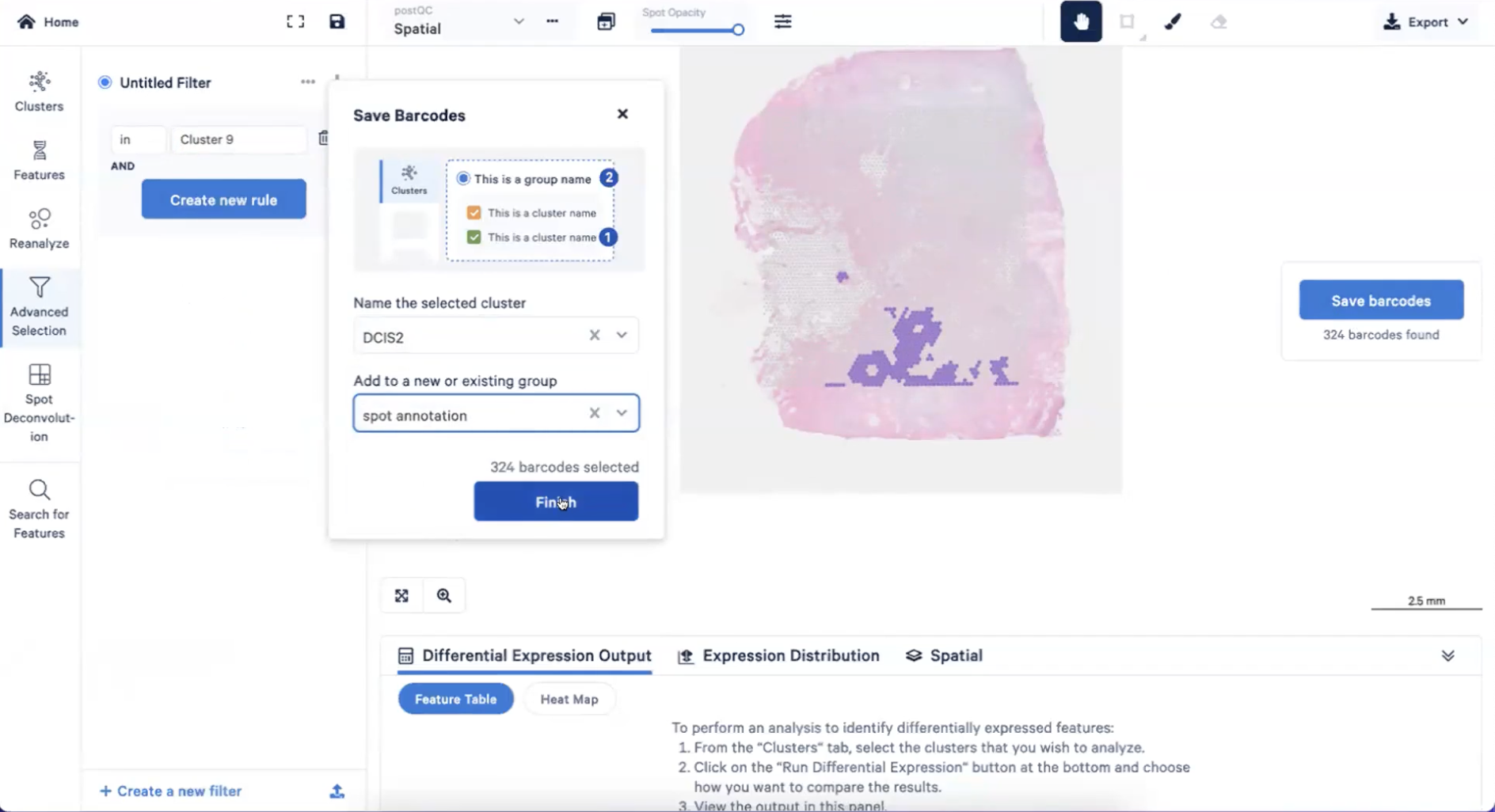
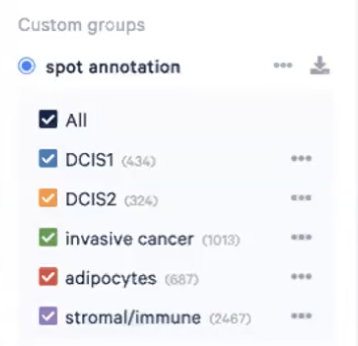
- 这样,我们就在cluster中出现了一个新组”spot annotation”,并且包含2种细胞DCIS1和DCIS2,如下图所示
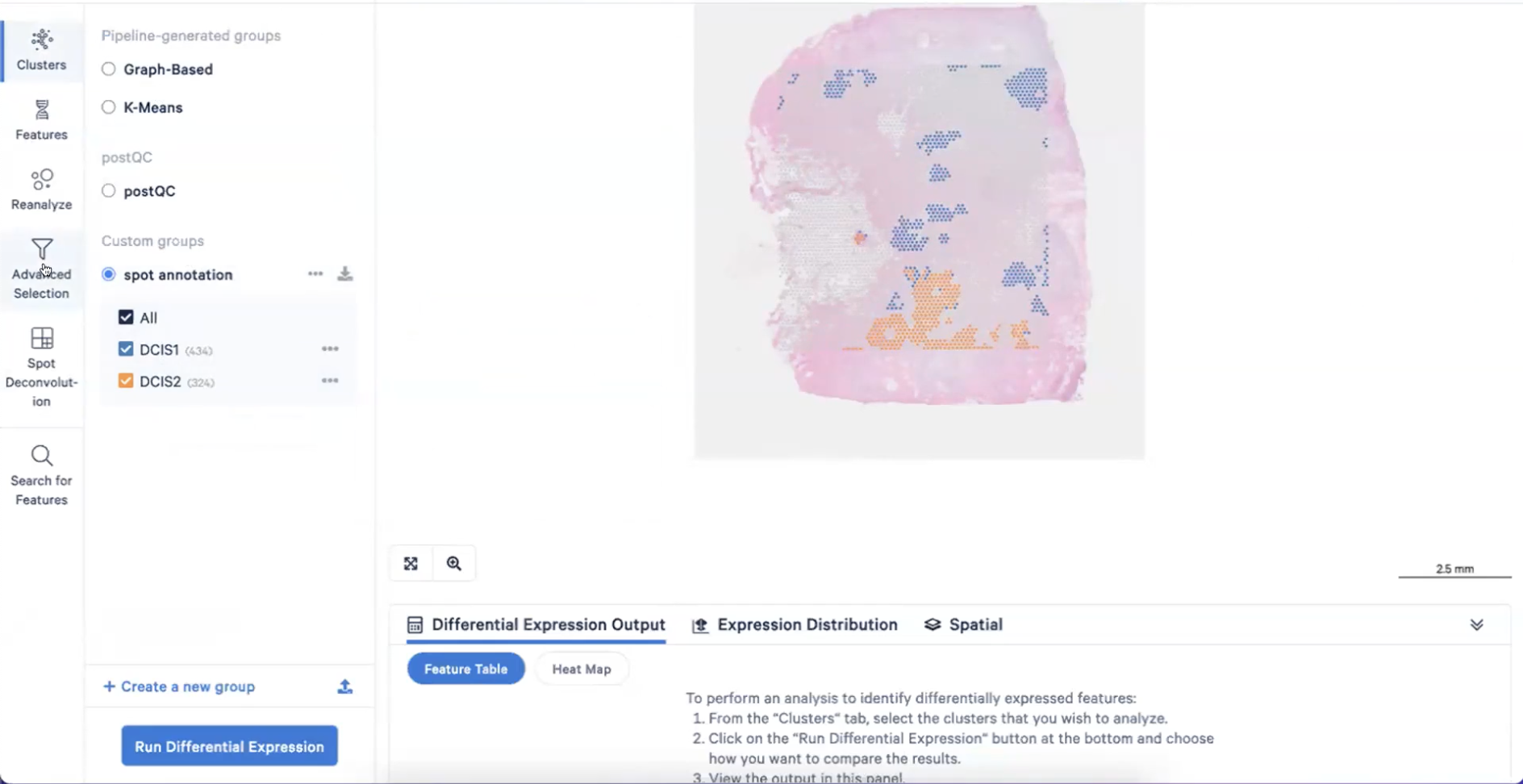
- 回到postQC,我们前面提到cluster4、7、8、9、11都是肿瘤细胞,而4和9已经被我们注释为导管原位癌了,剩下的7、8、11又不表达肌上皮的marker,因此我们可以用进展期导管腺癌去注释这些肿瘤细胞,也就是invasive cancer
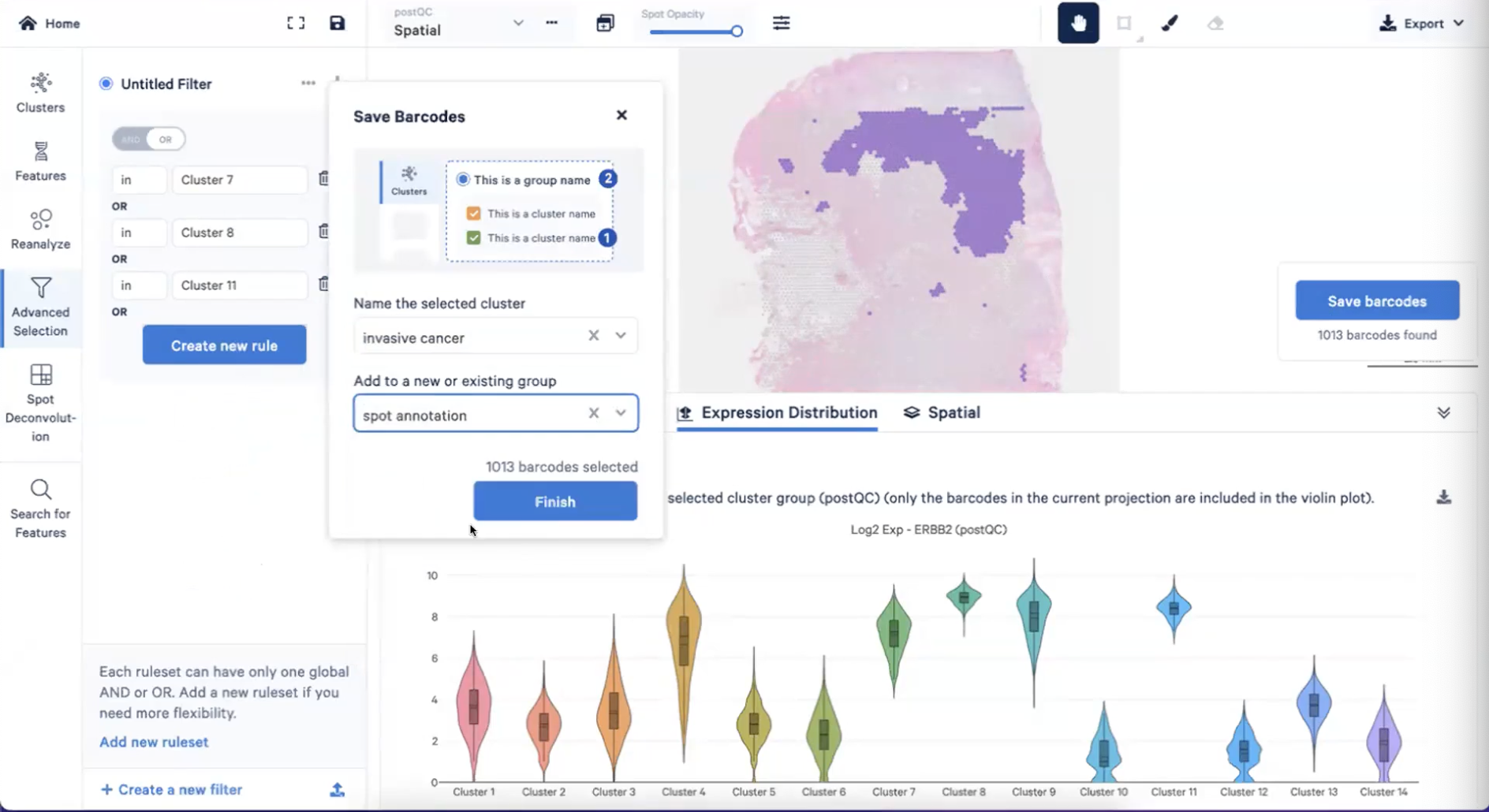
- 这时,我们发现切片的某个区域(下图的左下角)富含脂肪细胞,而cluster10、13和14三群细胞覆盖了这个区域,
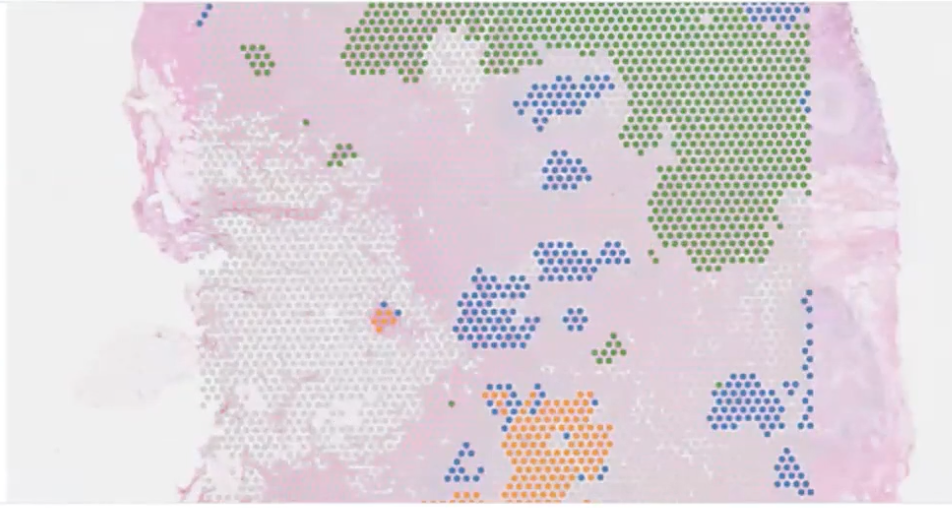
- 这些cluster高表达脂肪细胞相关的marker,ADIPOQ,分别间下图1-3
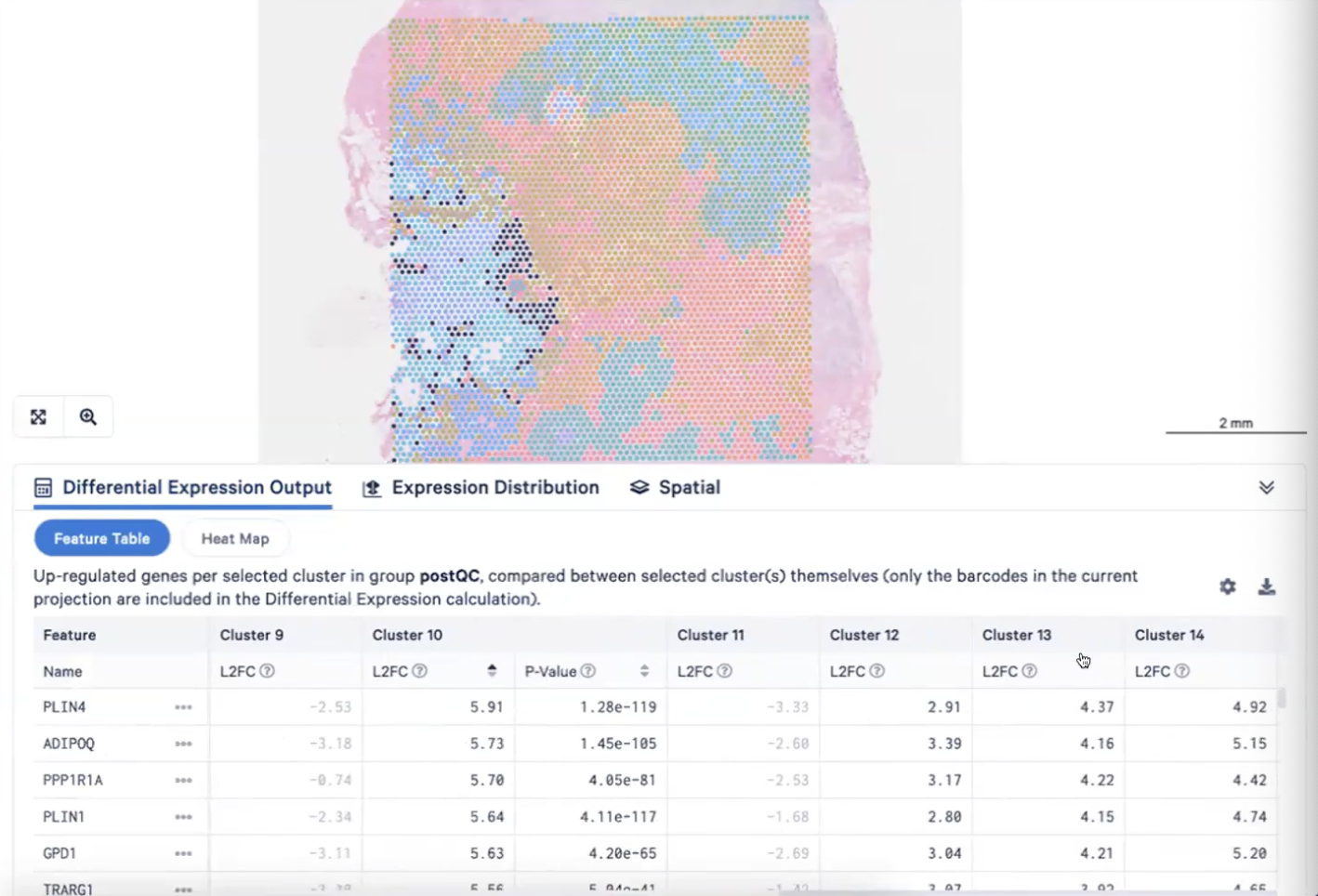
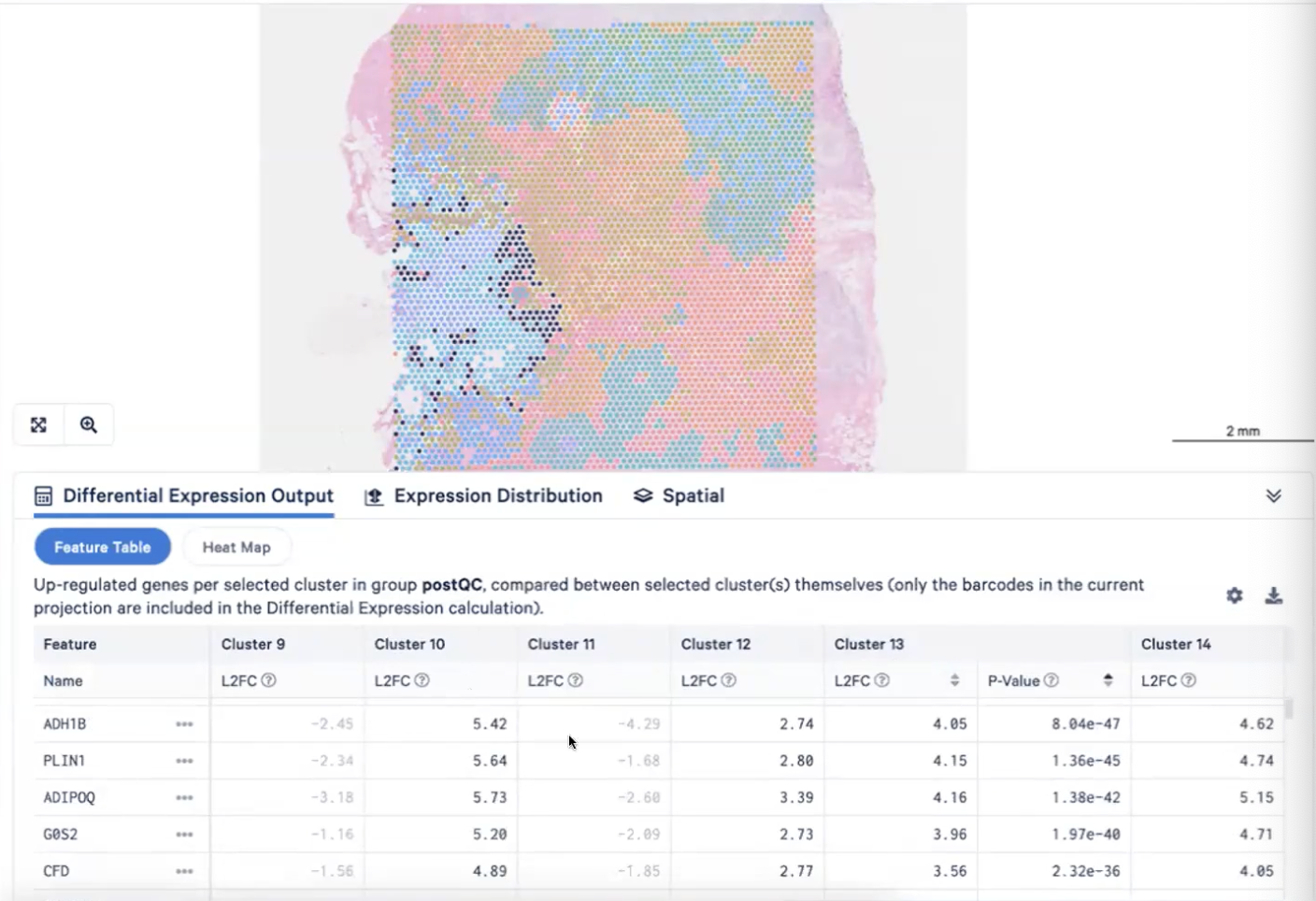
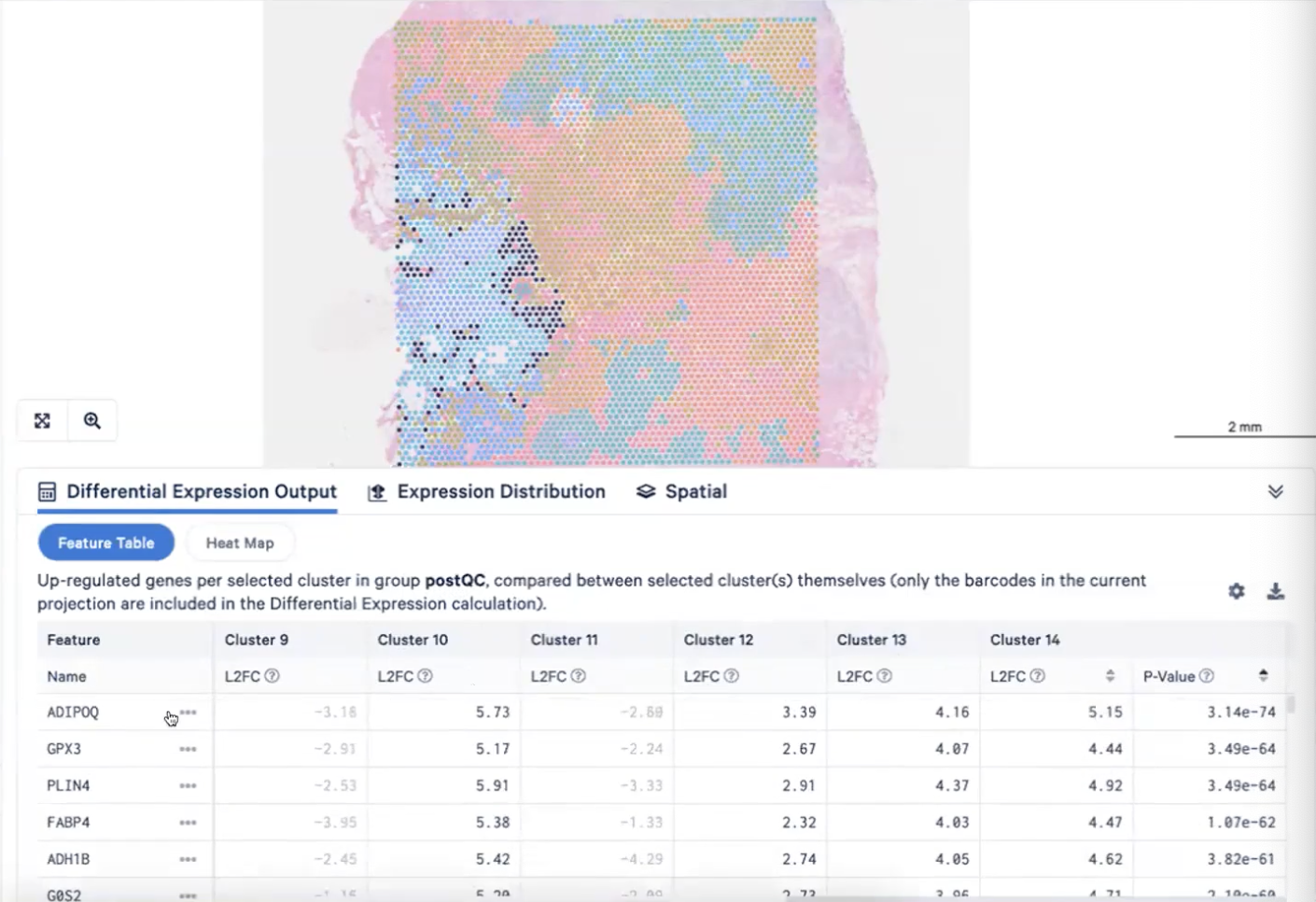
- 因此,我们可以把cluster10、13和14注释成脂肪细胞adipocytes
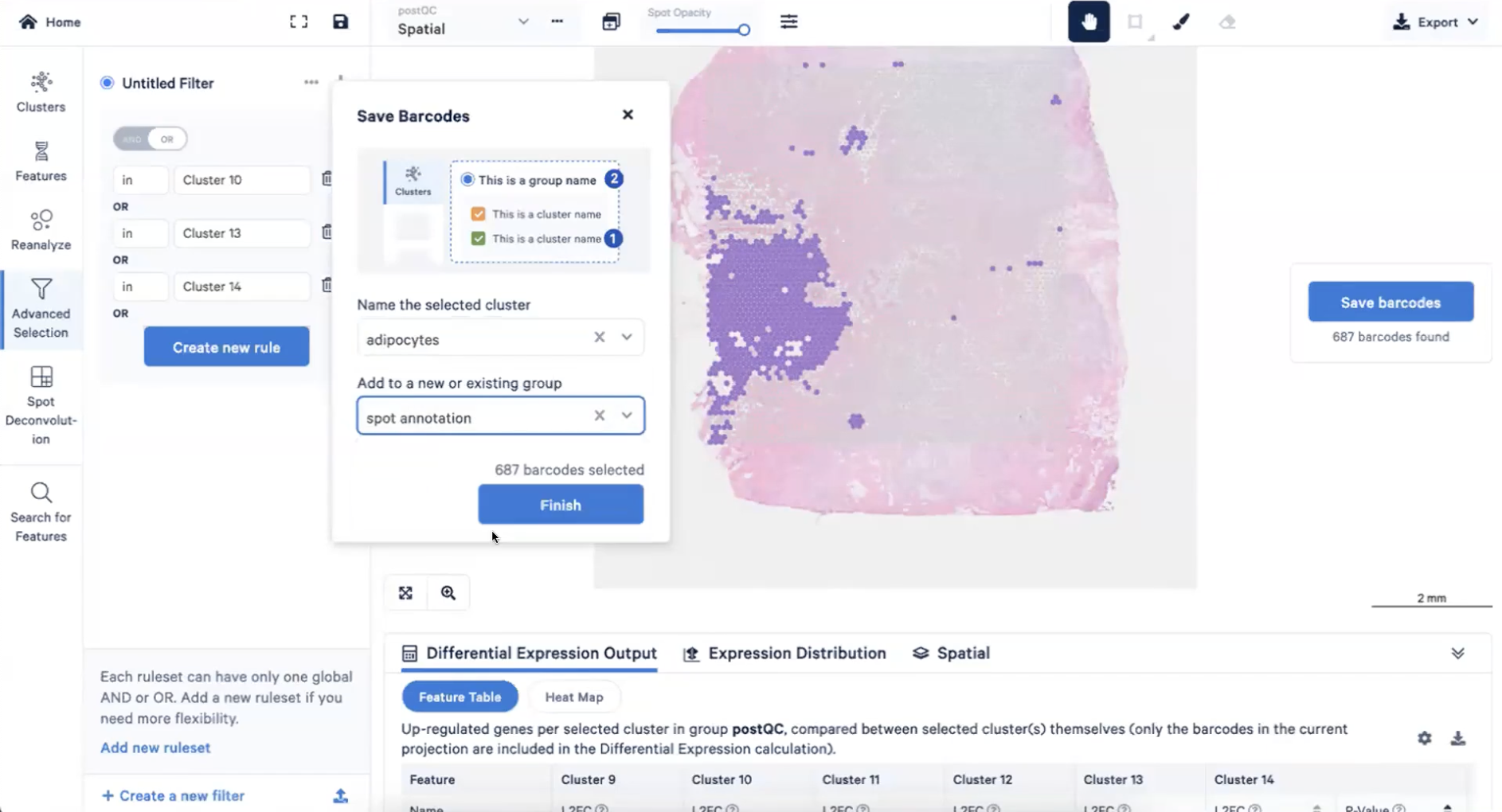
- 并且,剩下的细胞我们暂时想不到该如何进行手动的细胞注释,所以索性归到一类里,并且命名为”what are they”哈哈哈
- 想要知道这些细胞具体是什么,我们可以运行差异表达基因,去比较这群细胞和其他所有细胞相比,高表达哪些marker gene,得到的结果发现这些细胞中高表达间质和免疫的marker,所以我们可以把这群细胞命名为间质/免疫基因
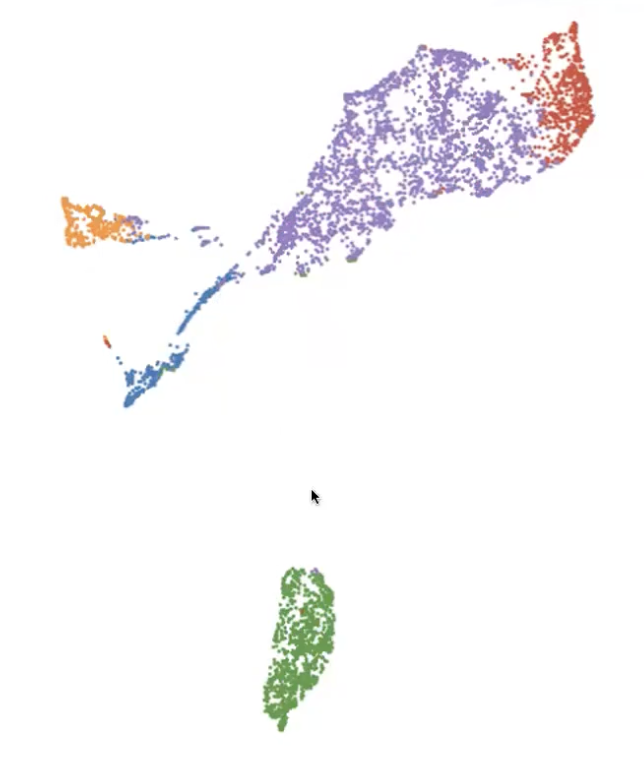
- 在UMAP图上,我们可以发现invasive cancer组的基因表达情况和其他细胞存在显著的差异,以至于它们自己形成了一个岛,剩下的细胞图例如上图所示;我们同样可以观察到两群早期肿瘤的基因表达情况存在着显著的差异,在UMAP上可以完全区分开
- 至此,我们可以顺利成章地提出新的科学问题“为什么这两群早期肿瘤的差别如此之大”,这个时候,由于Visium技术每个spot覆盖55μm,每个spot中可能存在2-4个细胞,我们无法准确地判断这些孔中的细胞组成和类型,如果我们能使用同样的组织产生单细胞数据(对于FFPE样本可以使用Flex单细胞技术),那我们就可以用单细胞数据进行reference-based cell-deconvolution,结果如下图所示,每个spot有多少细胞类型和对应的组成部份

- 我们可以看到之前高表达cluster 4的区域是高表达CPB1的DCIS1型细胞所在的地方,红色区域是invasive cancer所在的地方,而黄色区域内已经逐渐开始有红色细胞(下图),提示导管原位癌已经开始出现invasive cancer的形态了

- 相对的,在DCIS2型细胞中,几乎没有看到恶性程度更强的invasive cancer(下图),也就是说这群细胞几乎没有incasive cnacer

- 回到肌上皮,我们可以发现在高表达恶性肿瘤细胞的区域,肌上皮的表达是不完整的(肌上皮有两种,而恶性肿瘤细胞的区域只有其中一种),但相对的,DCIS2的区域表达的肌上皮就更完整一些,提示这两个区域的肿瘤存在差异
- 通过单细胞数据的整合,我们可以实现每个spot的反卷积,了解每个spot的组成部份,进一步放大组织上的细胞组成结构> 来源:,帮助我们更好地去了解组织形态


