
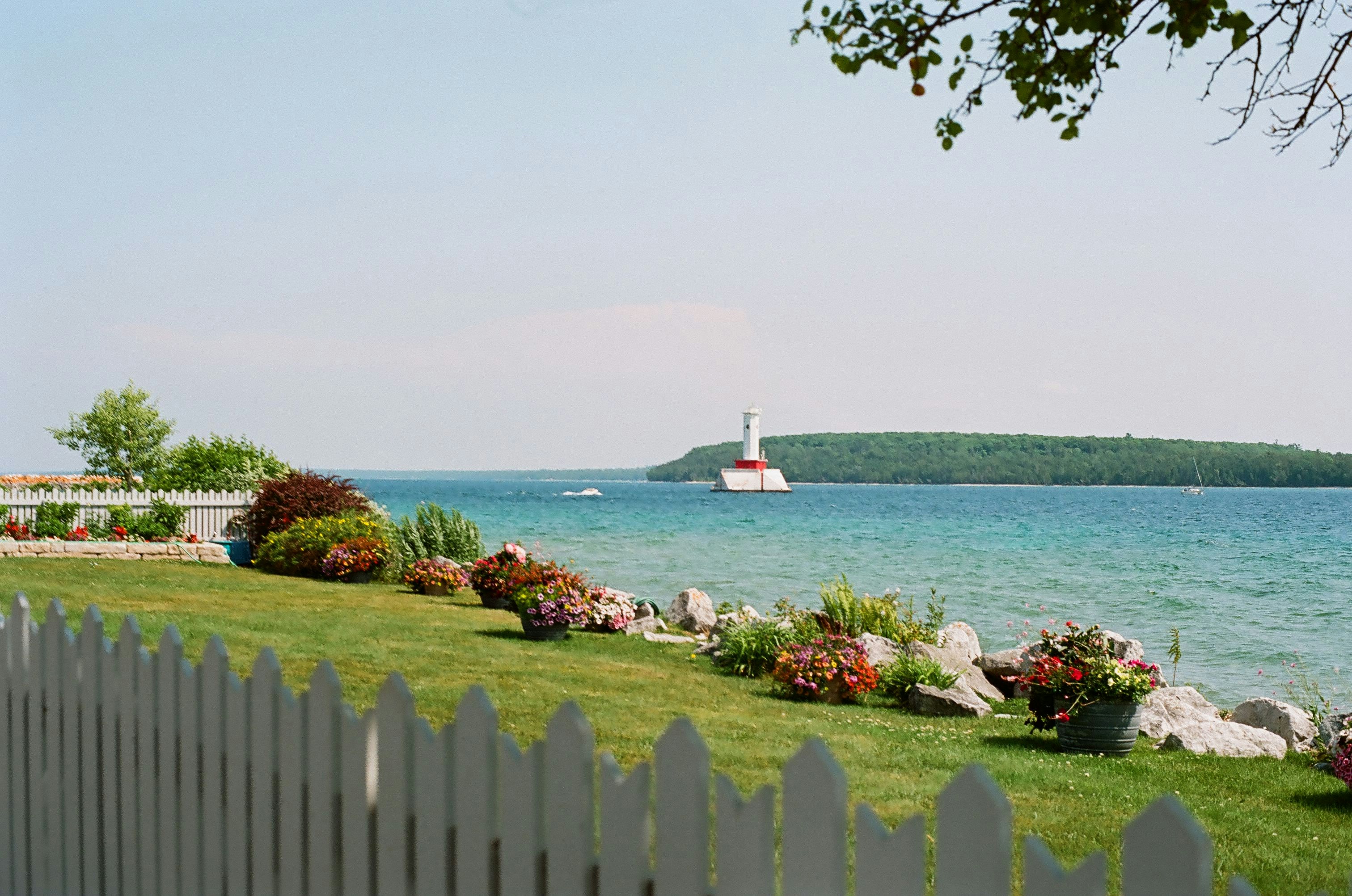

原机下载数据
- 数据分析从获得测序数据开始,一般我们拿到的二代测序下机数据为FASTQ格式,储存了测序序列,也就是序列信息(reads)以及对应的质量信息等,习惯上称之为raw data
- 复习Lesson 5中的内容
- FASTQ文件如图所示,每4行分为一个单元
- 第一行为序列名称,以“@”开头,一般是测序仪产生的,第三行也是序列名称,以“+”开头,为了节省储存空间,“+”之后的内容就不显示了
- 第二行是序列的碱基,也就是ATGC
- 第四行是测序的质量,每个字符都与第二行的同一个位置的碱基一一对应
- 如果是单端测序,对于每个样本我们只会得到一个这样的FASTQ文件;如果是双端测序,对于每个样本我们会得到2个FASTQ文件,一个是R1的,一个是R2的
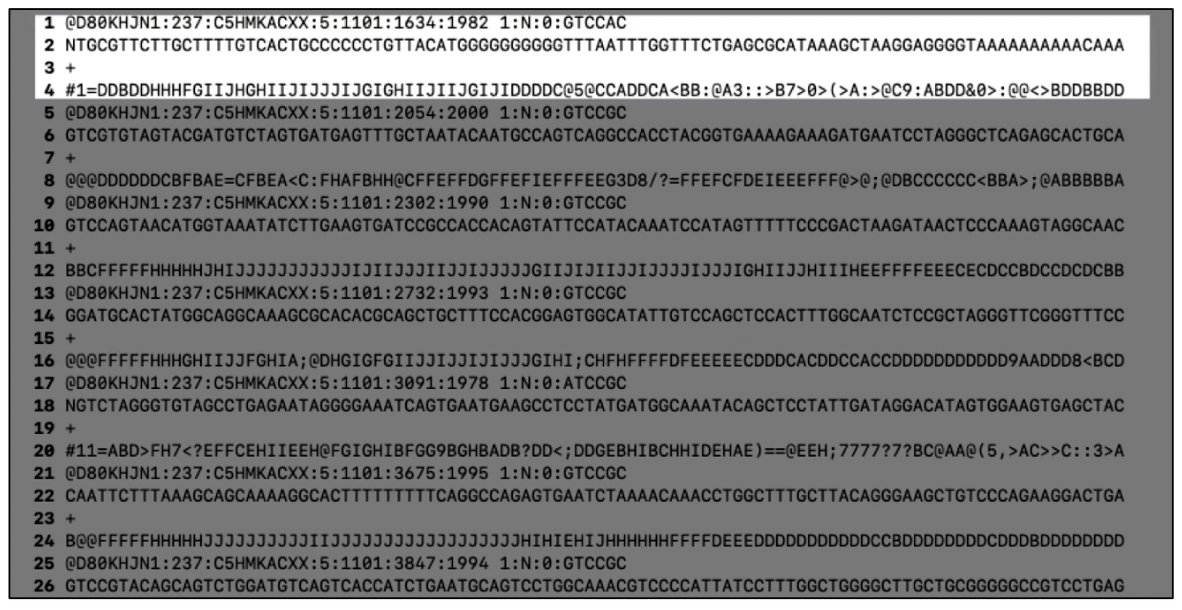
单端测序和双端测序的比较

Paired-end sequencing facilitates detection of genomic rearrangements and repetitive sequence elements, as well as gene fusions and novel transcripts.
In addition to producing twice the number of reads for the same time and effort in library preparation, sequences aligned as read pairs enable more accurate read alignment and the ability to detect insertion-deletion (indel) variants, which is more difficult with single-read data.
测序质量评估和质控处理
质量评估:FastQC
- FastQC官网:http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
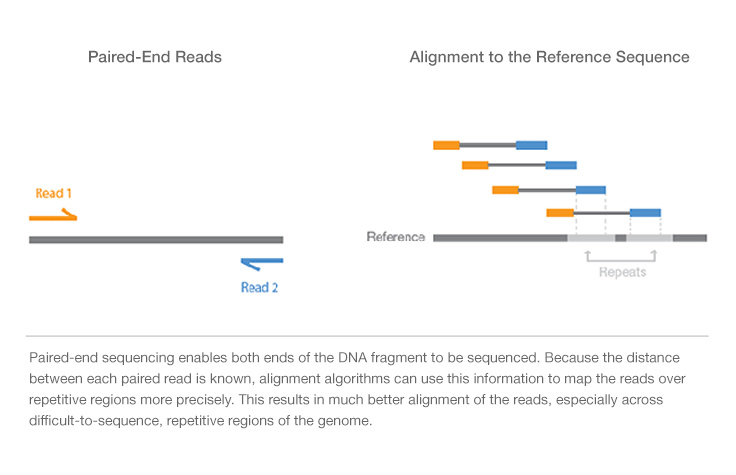
- 这个软件运行结束后会生成两个文件:一个是 html 网页文件,一个是zip压缩文件。如果我们使用浏览器打开 html文件,它显示的内容如下图所示
- 展示的内容
- 基本信息
- 序列整体测序质量统计
- 每条序列的测序质量统计和碱基分布
- 序列平均 GC含量分布图
- N含量统计
- 序列测序长度统计
- 重复序列统计
- 等等
- 一个合格原始数据,它的质控结果应该全是绿色的
碱基质量评估的结果
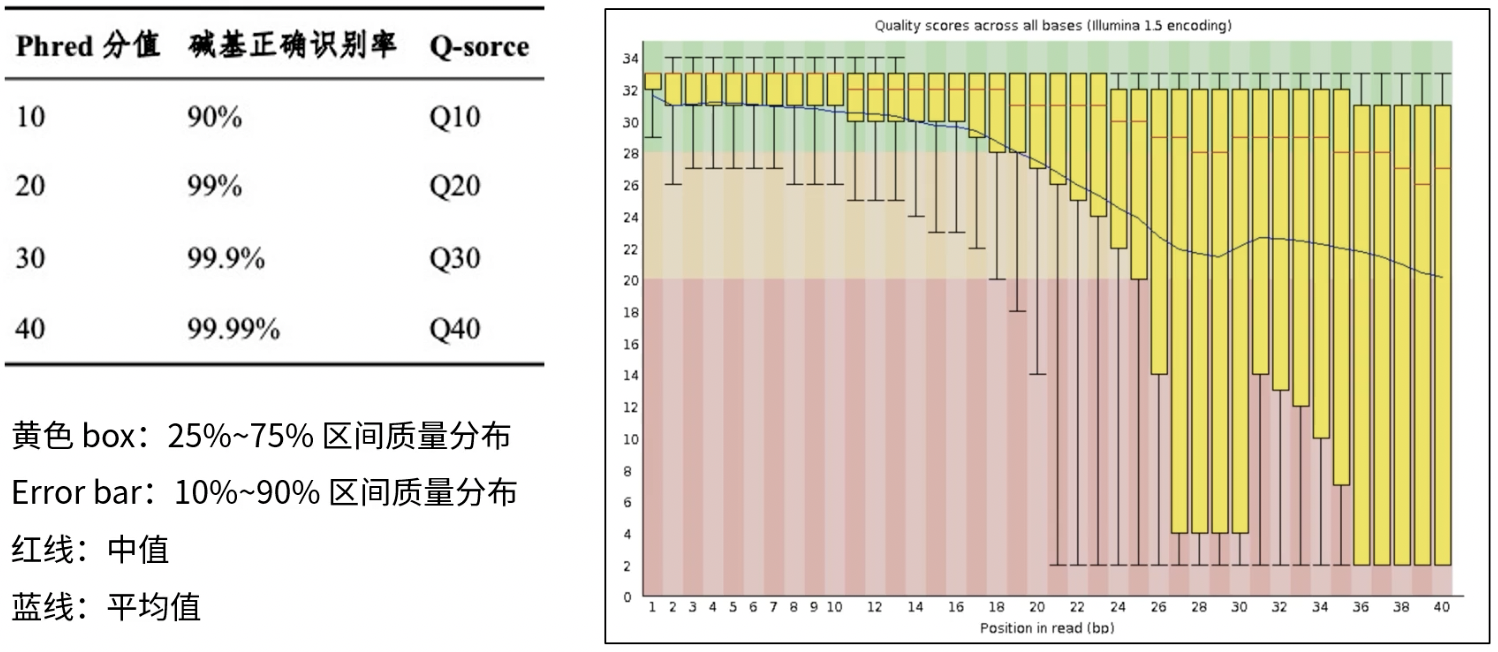
- 横轴为read的长度,纵轴为Phred数值,也就是质量得分,是通过概率模型得到的
- Phred=30时,碱基正确识别率为99%,这就是我们常提到的Q30
- 图片的判读
- 黄色box:25-75%区间的质量分布
- 黑色的error bar“10-90%区间的质量分布
- 红线:中位数
- 蓝色:平均值
- 如何根据一份数据的这张图对数据进行评估:我们希望所有位置的10%分位数的Phred值>20,即最下面的黑色横线要在黄色区域以内,才能认为质量合格
质控处理
- 质控常用软件:Trimmomatic、trim-galore、cutadapt
质控处理的目的
- 去除低质量碱基
- 去除低质量reads
- 去除接头序列
- 去除头部和尾部的:低质量的,以及过多的reads
- 丢掉小于一定长度的reads
- 经过质控处理,我们得到的仍然是FASTQ文件,我们把这样经过质控处理的文件称为clean data
基因组比对
- 比对常用软件:Hisat2、STAR、Tophat(前几年比较常用,现在被前两个替代了)
- 比对完成之后的输出文件是SAM文件,这是一种序列比对格式标准,是以TAB为分割符的文本格式,用于储存测序序列mapping到基因组上的结果,当然也可以表示任意的多重比对结果
SAM文件内容
- 注释信息和比对结果两部份组成
- 注释信息可有可无,都是以”@”开头的
- 比对结果部份表示一个片段的比对信息,一共包括11个必须字段和1个可选字段,字段之间使用tab进行分割
SAM对比示例

- 第一列:对比片段的ID
- 第二列:比对情况(第一行的163,这里每一个数字代表一种比对情况,这里的值是符合情况的数字相加的总和
- 第三列:对比到参考序列上的染色体号,如果没有对比上,这一列以” * “替代
- 第四列:read比对到的参考序列上的,第一个碱基所在的位置,如果没有比对上,这个值是0
- 第五列:对比的质量分数,这个值越高,说明这个read比对到参考基因组上的位置越唯一,如果是0,说明没有比对上,或者比对到了重复序列
- 第六列:简要的比对信息表达式,是以参考序列为基础,使用数字+字母表示比对的结果
- 图中示例的150M:这条read的150个碱基全部比对上
- 图中示例的3S6M1P1I4M
- 3S:前三个碱基被剪切去除了
- 6M:6个碱基比对上了
- 1P:打开了一个缺口
- 1I:有1个碱基插入
- 4M:最后4个碱基比对上了
- 第七列:read2比对上的参考序列的编号,这是针对双端测序而言的,单端测序这里是” * “
- 第八列:read2比对到的参考序列上的,第一个碱基所在的位置,如果没有比对上,这个值是0
- 第九列:片段的长度
- 第十列:序列片段的序列信息,即测序的read的序列信息,格式同FASTQ
- 第十一列:序列的质量信息
- 得到SAM文件后,我们通常会把它转换为BAM文件,BAM是SAM的二进制文件,储存的信息是一样的,但是占用的储存空间更加小
基因表达水平定量
- 一般使用HTSeq,本质是一个python库,具有多种功能,但我们只用其中的htseq-count来进行基因定量
- htseq-count的功能:根据基因结构注释文件(.gtf),对排序过的BAM文件进行基因reads数的统计,最终得到counts计数文件
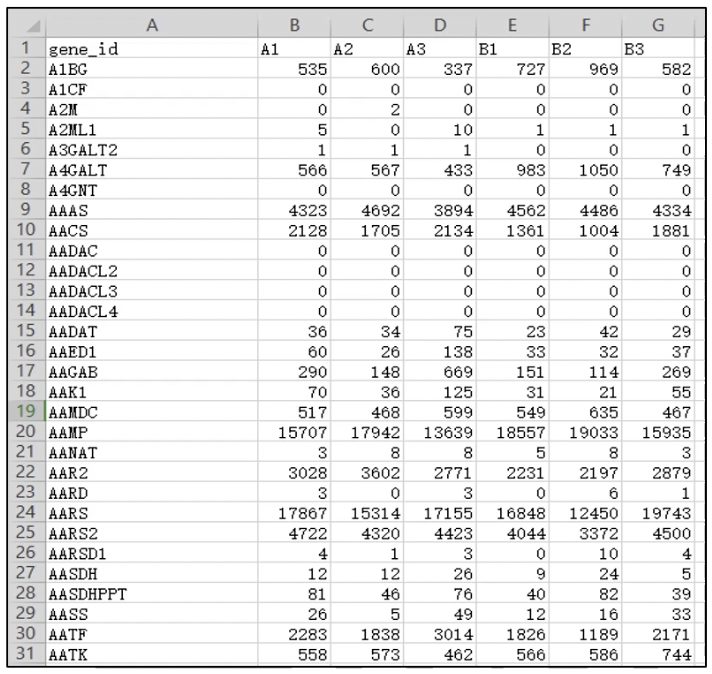
- 上图中展示的是一个整理过的 counts 文件,列是样本,行是基因名,数字代表这个样本中比对到每一个基因的reads的计数。
- 如果是单端测序,那么一个read比对到基因 A上,计数就为 1,如果是双端测序,一对 reads比对到基因A 上,计数也为1
- 一个样本会得到一个counts文件,这里展示的是将多个样本整理到一个文件中的counts。counts是比对到基因上reads 的绝对计数也是下一步进行差异分析的输入文件。
差异基因分析
差异分析常用工具
- DESeq2
- edgeR
- limma
差异分析流程图
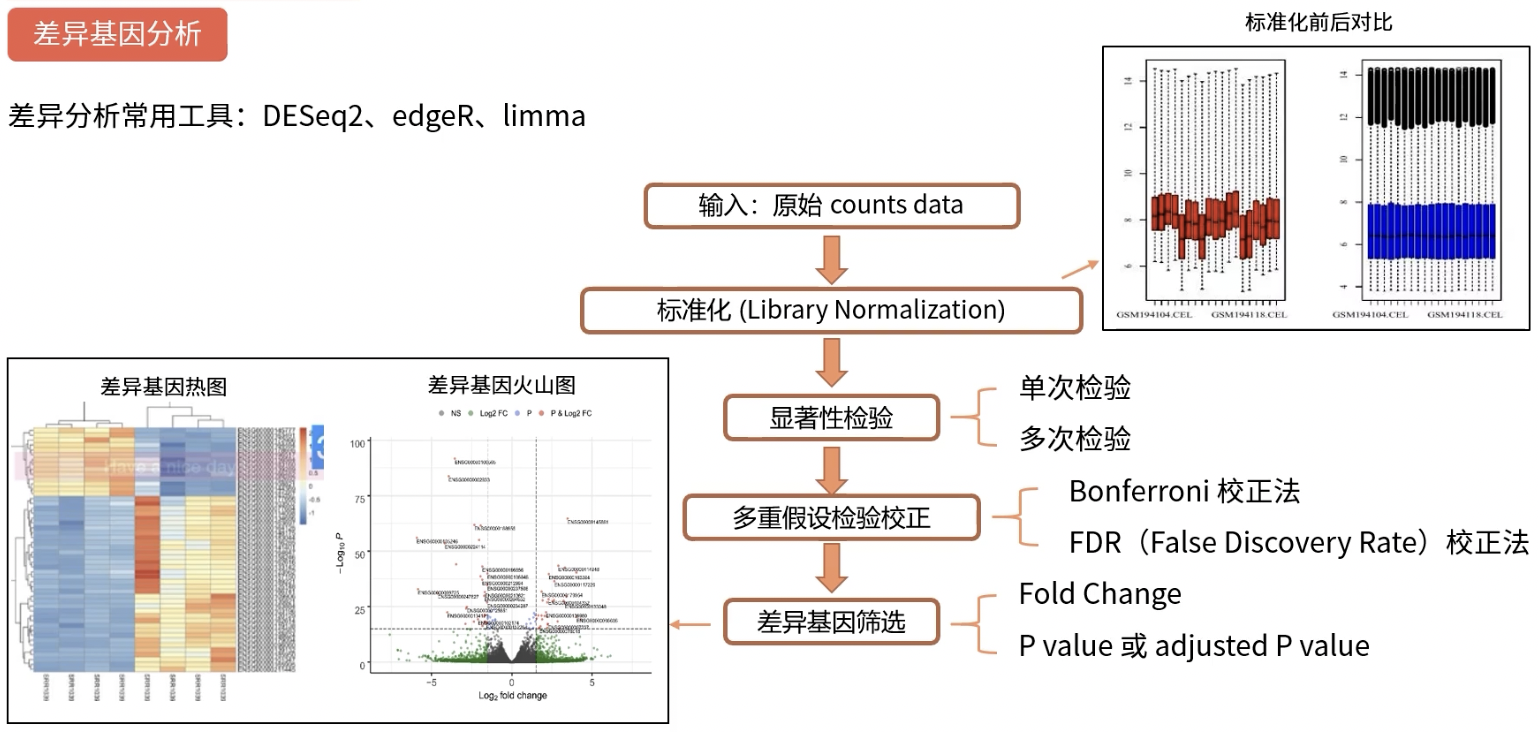
- 多重假设检验校正:Bonferroni校正法较严格,FDR校正法较温和
- 常用的筛选条件
- p value<0.05
- adjusted p value<0.05是更加严格的做法
- Fold Change≥2
扩展:其他的定量数据
定量数据的类型
- Counts:对比到参考基因组上的Reads(Fragments)的原始计数
- RPKM/FPKM:每百万个reads/fragments中来自某基因每千碱基长度的reads数,即将比对到基因的read数除以比对到基因组上的所有 read 数与基因的长度
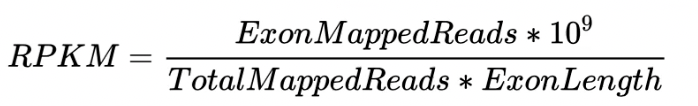
- Reads与Fragments的关系:reads是指下机后fastq数据中的每一条 reads,fragments呢是指每一段用于测序的核酸片段,在单端测序中,一个fragments只测一条reads,reads数与fragments数目相等;在双端测序中,一个fragments测两端,会得到2条reads,这2个reads只能算一个fragments,所以fragments的值是reads的1或2倍
- TPM:消除了exon长度造成的差异
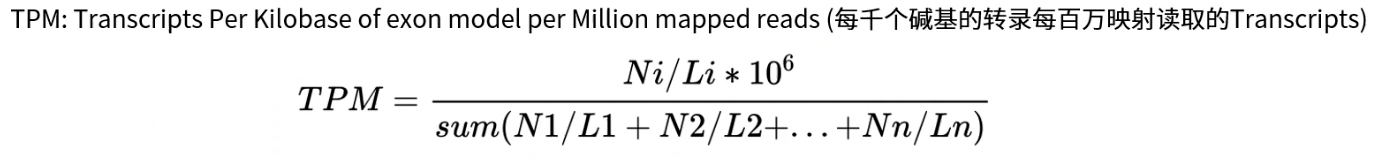
- RPM/CPM:比对到某一个基因的read/count数除以基因组上总的read/count数,有助于样本之间进行比较
