测序技术预备知识
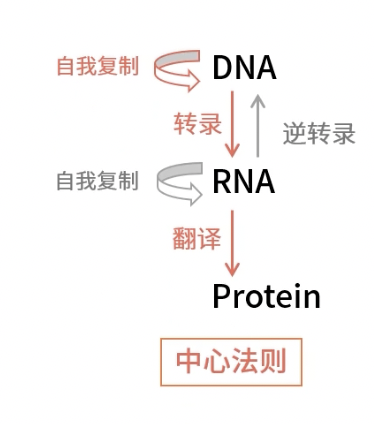
- 中心法则
- 常见组学
- DNA层面的是基因组学 Genomics
- DNA的甲基化、组蛋白修饰层面的是表观遗传组学epigenomics
- RNA层面的是转录组学transcriptomics
- 蛋白质层面的叫做蛋白质组学proteomics
- 代谢组学:对体内的代谢产物同时进行定性和定量分析的研究,主要针对的是分子量 1000以内小分子物质
测序原理
一些基础概念:
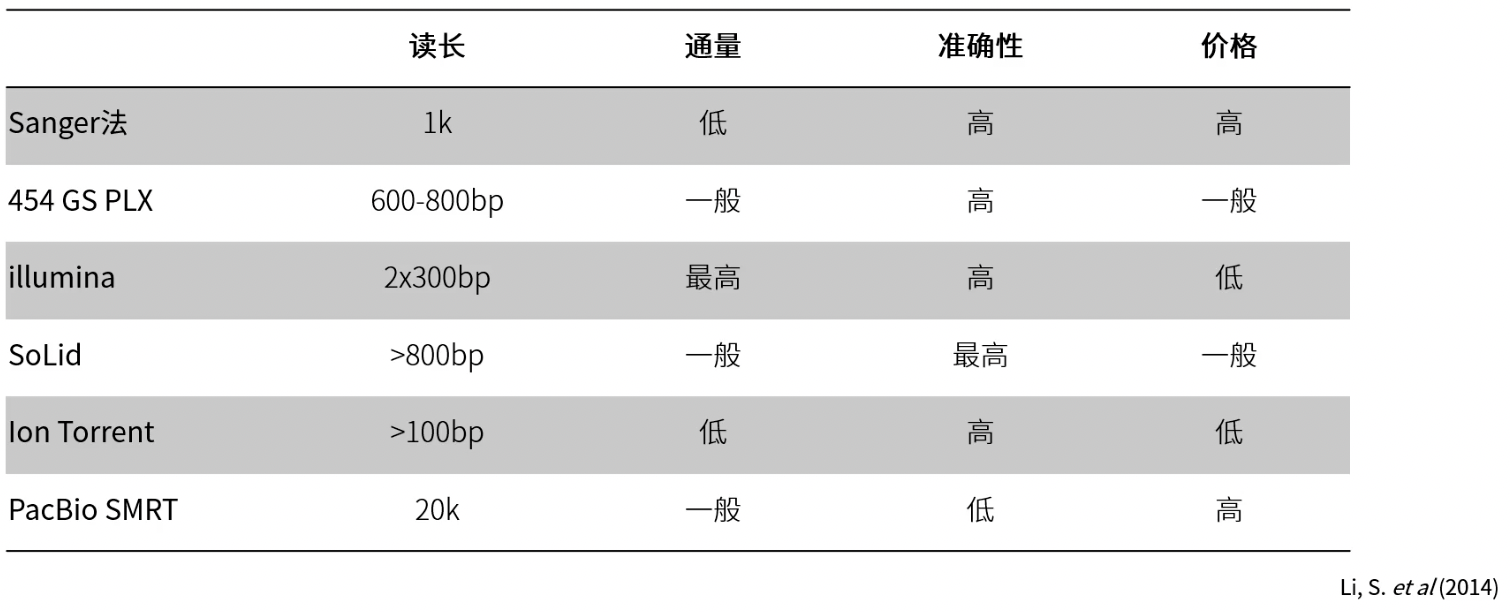
- 读长:测序反应所能测得序列的长度,不同的测序平台所能获得目的序列的长度各不相同,当待测序列的长度超过测序仪的最大读长时,得到的结果准确性会大幅降低。
- 通量:单位时间内所能产生的数据量。
- 接头(Adaptor):特定的一段DNA序列,在构建测序文库时连接在片段化的DNA末端,与测序槽中固定的接头序列匹配,从而固定待测序列在测序槽中的位置。
- 测序文库:高通量测序技术具有测序读长的限制,因此在进行测序之前,需要将提取得到的样品DNA打断成为符合测序仪器读长的小片段,经过片段长度筛选、添加接头和定量,即构成了能够用于高通量测序的DNA文库。
- 单端测序(Single-end):在构建DNA文库时,将测序引物连接在DNA片段的一端,然后在末端添加接头序列,在进行上机测序时只能从序列的一端开始进行测序。
- 双端测序(Paired-end):在构建DNA文库时,在DNA片段的两端均连接测序引物和接头,在进行上机测序时可以分别从序列的两端进行测序,从而使得测序读长增加为原本的两倍左右。
- 测序深度:测序得到的总数据量与待测基因组大小的比值,假设待测基因组大小为100Mb,测序深度为30X,那么最终得到的数据量为3G。
- 质量控制:按照指定的标准对测序得到的序列进行筛选,去除不合格序列的过程。
- Raw data:测序仪下机得到的原始数据。
- Clean data: 原始数据经过质量控制后,得到的可以用于后续分析的数据
一代测序:Sanger测序法
- 原理:链终止DNA测序法
- 实验步骤
- DNA碎片化
- PCR扩增与体外克隆
- ddNTP法循环测序
- 凝胶电泳获得序列
- 优点
- 读长较长,600-700bp
- 准确率高,99.9%+
- 缺点
二代测序
- 原理
- Roche/454的焦磷酸测序
- ABI/SOLiD的连接法测序法
- Illumina/Solexa的边合成边测序
- 优点
- 缺点
- 读长较短:原因在于测序长度越长,杂信号越多
- PCR富集序列过程中可能丢失序列或者引入错配碱基
- 常用的二代测序种类
- 全基因组测序(Whole Genome Sequencing, WGS):全部基因进行高通量测序
- 全基因组关联分析(Genome wide association study,GWAS):研究某一特定人群中研究遗传突变和表型之间的相关性。
- 全外显子测序(Whole-exome sequencing, WES):全部外显子区域的总和进行高通量测序
- 转录组测序(RNA sequencing, RNA-seq):所有的RNA进行的高通量测序
- ATAC-seq(Assay for Targeting Accessible-Chromatin with high-throughout sequencing):处于开放状态的染色质区域进行的高通量测序
- ChIP-seq(Chromatin Immunoprecipitation sequencing):研究体内蛋白质与DNA的相互作用单细胞测序(Single-cell sequencing):单个细胞水平上对基因组进行扩增和测序
全基因组测序
- 概念:对个体进行全基因组范围的高通量测序,并在个体或群体水平上进行差异性分析的方法。
- 用途:在全基因组水平上全面挖掘DNA水平的遗传变异,包括单核苷酸变异(SNV)、插入/缺失(InDel)、拷贝数变异(CNV)和结构变异(SV)等多种全面的突变信息,包括编码区和非编码区,可用于:常见疾病易感基因检测、遗传病基因检测、肿瘤基因组检测、个性化用药检测等。
- 优势
- 数据量大,全面扫描基因组上的变异信息,一次性挖掘大量的生物标记物,
- 准确性高、可重复性好、定位精确,已被广泛应用于疾病、癌症的基因组研究。
- 缺点:
- 测序会获得大量无效信息,需要进一步挖掘分析,
- 主要是基因组层面的信息,不包含转录组。
- 大致价格(以公司报价为准):8千-1万
全基因组相关分析Genome wide association study,GWAS
- 概念:研究某一特定人群中研究遗传突变和表型之间的相关性,比如研究单核苷酸多态性(SNPs)与主要疾病等性状之间的关联。
- 用途:常用于病例对照研究,将患者的全基因组范围内检测出的SNP位点与对照组进行比较,找出所有的变异等位基因频率,为复杂疾病研究提供更多线索。
- 优势:GWAS分析建立在单碱基水平,速度快,节约时间
- 缺点
- 基于严格的统计水准,可能损失潜在易感位点
- 目前主要关注SNPs,忽略了其他的遗传变异,对低频率的SNP发现不足(可以通过加大样本量和meta分析来解决)
- 着重发现新的位点,忽略了生物学功能研究
- SNP数据库不全,有赖于深度测序研究
- 大致价格(以公司报价为准):2千-5千
全外显子测序Whole-exome sequencing, WES
- 概念:针对全部外显子区域的总和进行高通量测序(外显子组约占全基因组的1-2%)。
- 用途:主要用于识别和研究与疾病相关的编码区的基因组变异,可以更好地排除无害突变及解释变异信息之间的关联和致病机理。
- 优势:
- 直接对蛋白编码序列进行测序,找出影响蛋白结构的变异;
- 高深度测序,可发现常见变异、低频变异及罕见变异;
- 性价比高,有效降低费用,周期和工作量。
- 缺点:数据量远不如全基因组测序
- 大致价格(以公司报价为准):2千-5千
转录组测序RNA sequencing, RNA-seq
- 概念:针对特定组织或细胞在某一发育阶段或功能状态下转录出来的所有RNA的集合进行的高通量测序,主要包括mRNA和非编码RNA。
- 用途:观察基因剪接转录、转录后修饰、基因融合、突变/snp和基因表达随时间的变化,或不同组间的基因表达差异。RNA-Seq的最新进展还可用于单细胞测序。
- 优势:
- 任意物种的全基因组分析,无需特异性引物
- 检测范围广,敏感性高,无需技术重复
- 性价比高
- 缺点:
- 存在误差,可以通过增加生物学重复来减少误差
- 分析结果需要实验验证(几乎所有生物信息学得到结果所必须的)
- 大致价格(以公司报价为准):1千-3千
ATAC-seq
- 全称:Assay for Targeting Accessible-Chromatin with high-throughout sequencing,通过转座酶Tn5容易结合在开放染色质的特性,对Tn5酶捕获到的DNA序列进行高通量测序。
- 开放染色质(open chromatin)是指DNA的致密高级结构变为松散的状态。 开放染色质有足够的区域允许一些调控蛋白(比如转录因子)与之相结合。DNA复制,基因转录都发生在开放染色质。
- 用途:全基因组范围内检测染色质的开放程度,可以得到全基因组范围内的蛋白质可能结合的位点信息,一般用于不知道特定的转录因子,用此方法与其他方法结合筛查感兴趣的特定调控因子。
优势:
- 样本量需求少,500-5万,比其他实验百万级别的需要量要小很多
- 对测序深度要求更低,性价比更高
- 一次性获得全基因组上的染色质开放区域缺点:适用面较窄
- 大致价格(以公司报价为准):1万-2万
ChIP-seq
- 全称:Chromatin Immunoprecipitation sequencing
- 概念:ChIP实验 (Chromatin immunoprecipitation)即染色质免疫沉淀,根据DNA与蛋白质相互作用的原理,分离富集与感兴趣的蛋白相互作用的DNA。ChIP-Seq即对分离得到的DNA扩增测序。
- 用途:明确知道感兴趣的转录因子是什么,根据感兴趣的转录因子设计抗体去做ChIP实验拉DNA,验证感兴趣的转录因子是否与DNA存在相互作用。
- 优势:揭示特定转录因子或蛋白复合物的在DNA上的结合区域,实际是研究DNA和蛋白质的相互作用,利用抗体将蛋白质和DNA一起富集,并对富集到的DNA进行测序。
- 缺点:ChIP需要专门的抗体,数据的质量依赖于抗体的质量。
- 大致价格(以公司报价为准):2千-5千
单细胞测序Single-cell sequencing
- 概念:对单一细胞的基因组或转录组进行测序,可以理解为单细胞水平上的测序。
- 用途:传统测序方法所展示的信息也是在多细胞水平上的平均信息,而单细胞水平上的测序则完全可以反应同一个细胞群里不同细胞的基因组和转录组状况。还可以分析稀有的细胞,特别是特定时空环境下的细胞。比如从环境中取样的微生物、受精胚胎、循环肿瘤细胞等。
- 优势:从细胞群体的研究到单个细胞的研究,可用于单细胞多组学研究和单细胞的功能状态研究,创建细胞发育谱系的分子图谱。
- 缺点:成本较高。
- 大致价格(以公司报价为准):2万-5万
三代测序
- 第三代测序在 DNA测序时,不需要经过 PCR扩增,实现了对每一条 DNA分子的单独测序,具有更长的读长
- 第三代测序技术仍处于研发阶段,目前费用相对高昂,应用面较窄,可以用于ctDNA测序、单细胞测序等。
- 特点:单分子测序+更长的读长长度
- 优点
- 缺点
- 准确率相对低,需重复测序以纠错
- 成本较高
- 生信软件较少
代表性检验平台
二代测序平台
GS FLX 454
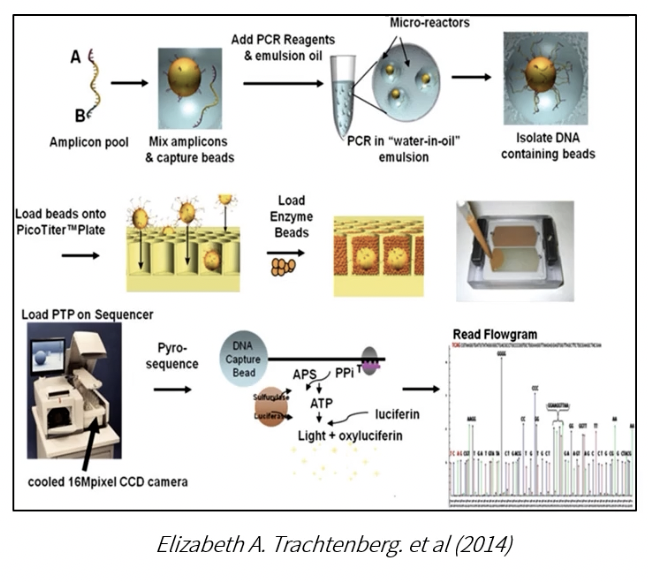
- 454 Life Sciences/ Roche于2004年推出了GS FLX system,是第一款商业型NGS测序仪器,现在已经淡出一线了
- 基本原理:一个片段=一个磁珠=一条读长,DNA片段无需进行荧光标记,无需电泳,边合成变测序,碱基在加入到序列中时,会脱掉一个焦磷酸,通过检测焦磷酸来识别碱基,因此也被称为焦磷酸测序。
- 扩增方式:常规PCR
- 测序方式:Pyrosequencing(焦磷酸测序)
Illumina
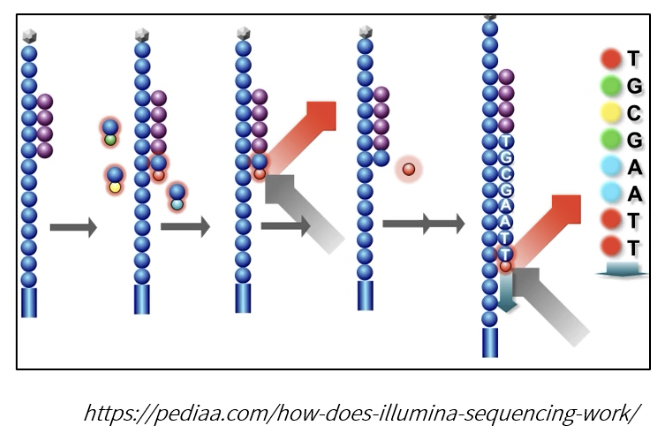
- Illumina最早于2006年推出了测序仪器Solexa和Hiseq系列,现在是使用最广泛的测序平台
- 扩增方式:桥式PCR
- 测序方式:Sequencing by Synthesis,即边合成边测序
SOLiD
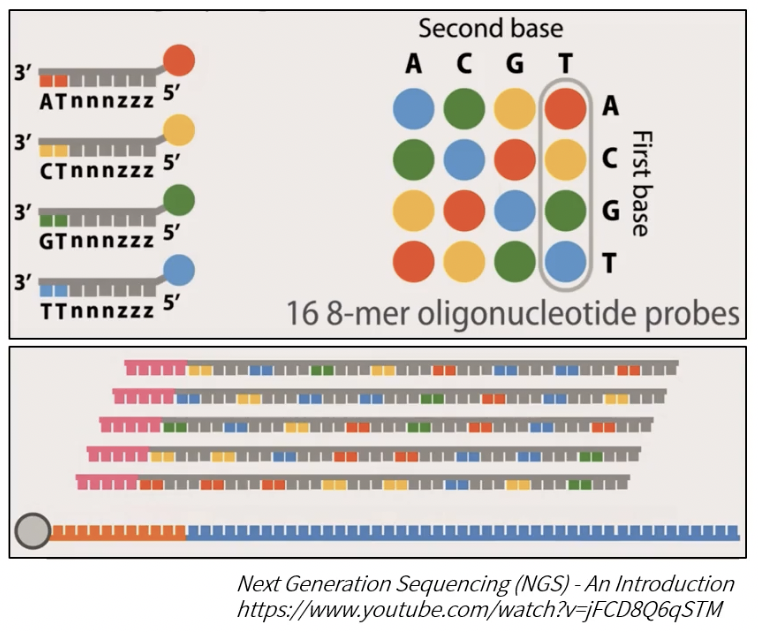
- SOLiD是ABL公司最早于2007年推出的测序仪
- 扩增方式:乳液PCR
- 测序方式:Sequencing by Ligation,连接法测序
Life Technologies
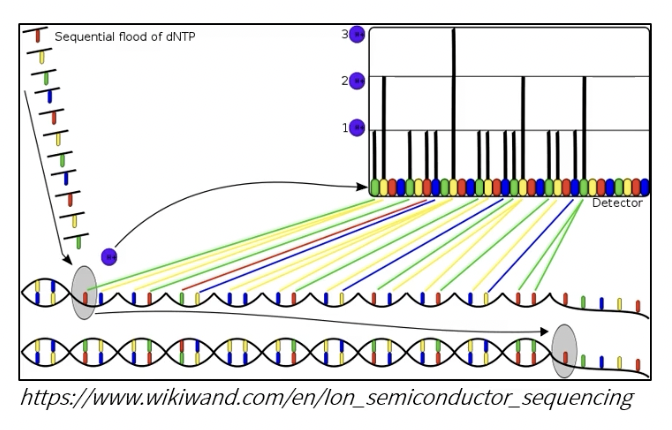
- Ion Torrent系列是Life Technologies公司最早于2010年推出的测序仪器
- 扩增方式:常规PCR
- 测序方式:离子半导体测序ISS
- 这个技术使用了一种布满小孔的高密度半导体芯片,在芯片上,一个小孔就是一个测序反应池。当 DNA 聚合酶把核苷酸聚合到延伸中的 DNA链上时,会释放出一个氢离子,反应池中的 PH发生改变,位于池下的离子感受器感受到氢离子信号,将氢离子信号直接转化为数字信号,从而读出DNA序列。这个方法的通量并不高,适合小基因组和外显子验证的测序。
三代测序平台
PacBio SMRT
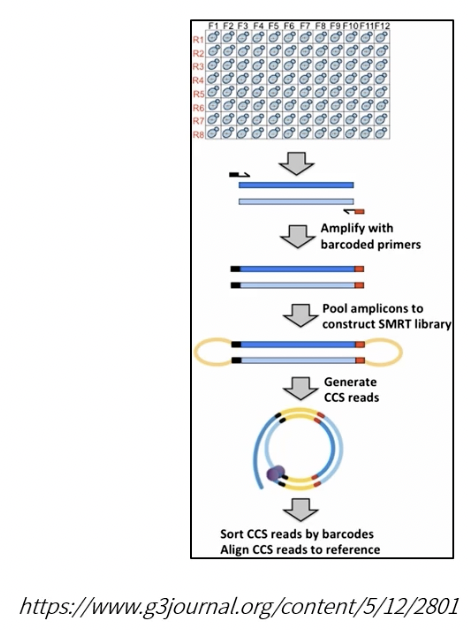
- 原理:边合成边测序,并且以SMRT芯片作为测序的载体
- 优点
- 缺点:错误率较高,需要通过多次测序来纠正错误
Oxford Nanopore
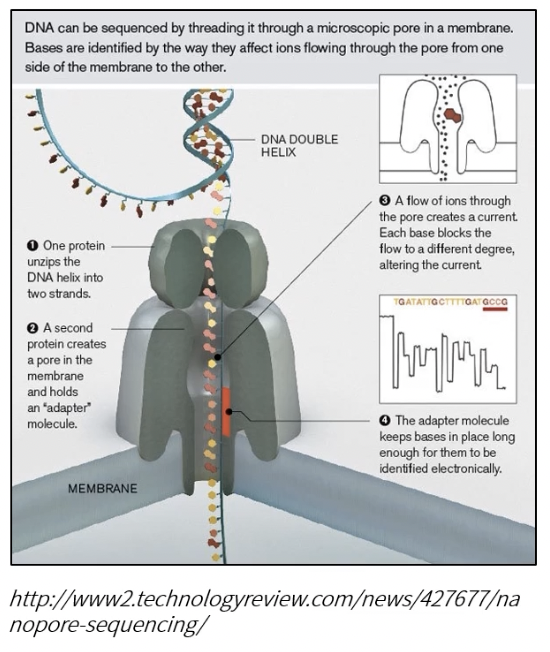
- 原理:纳米孔+电流检测技术
- 优点
- 读长很长
- 数据可以实时被读取
- 通量很高
- 样品制备简单而便宜
- 缺点:
- 错误率较高(1-5%),精准度较低
- 使用了水解测序法,不能重复测序
常见测序平台的对比
序列文件简介
FASTA格式
- 原始序列文件(如ab1、SRR、BCL等)通常首先转换成FASTA/FASTQ文件进行后续的处理,seq格式文件可以直接用软件分析,但是文件本身的信息准确性较差
- FASTA格式是BLAST组织数据的基本格式,无论是数据库还是查询序列,大多数情况都使用FASTA格式,后缀名一般是.fa
- 通常第一行以”>”开头,相当于注释,可以添加任意文本,通常标明样品编号,出处(数据库)等
- 第二行开始是序列正文,既可以是DNA序列,也可以是氨基酸序列,惯例使用大写,而且每行不超过80个字符,不符合规定的字符会被忽视
- 有时以” * “结尾
- FASTA格式允许将多段信息合并到一个FASTA文件中,各段序列凭借序列前的”>”行区分开

FASTQ格式
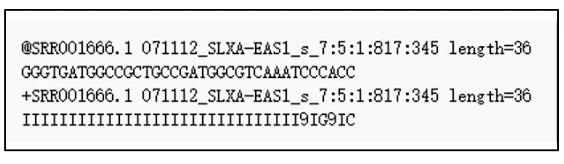
- 保存测序仪得到的核酸序列,以及其测序质量信息的标准格式,每条序列由4行文件组成
- 第一行由“@” 字符开头,其后是(Ilumina,NCB/等)序列ID,再后与FASTA文件第一行相同,是补充描述
- 第二行序列正文。其中无法判断的碱基用N表示
- 第三行以“+”字符开头,其后可跟序列ID,或直接省略内容
- 第四行按与序列正文对应的顺序,标记各碱基测序质量,按照以下符号按照从左到右的顺序标志,由低到高的质量得分

- 一些举例的测序图片

SAM/BAM
- SAM(Sequence Alignment Map)是一种用来存储从reads到参考序列的比对信息的文件格式,是得到序列文件(FASTA/FASTQ)后继续进行数据分析时会涉及到的一种数据类型。SAM 文件由注释头和比对结果两部分组成。
- 比对结果:每一行是一个read,包含11个必须字段和任意多个可选字段,字段间用制表符(tab)隔开。
- 前11个字段所包含信息为
- QNAME: 比对片段的read name,通常包括测序平台等信息。
- FLAG:位标识,一个数字表征一种template mapping情况。
- RNAMW:参考序列的编号。
- POS:比对到的位置(从1开始计数;若无匹配记为O)。
- MAPQ:mapping的质量打分,越高说明read的mapping越特异。
- CIGAR:简要比对信息表达式。
- RNEXT:配对的reads比对到的参考序列名称。
- PNEXT:配对的reads比对到的位置。
- TLEN: reads与配对reads组成的片段的长度。
- SEQ:reads序列。
- QUAL:reads质量值(格式同FASTQ一样)。
- BAM (Binary Alignment Map)是将SAM信息转换为二进制格式存储的文件,对存储空间的要求更小,检索更快。