

基本介绍
- NetworkAnalyst是一个基因表达谱和荟萃分析的可视化在线分析平台,集成了先进的统计方法和创新的数据可视化系统
- 可以支持:高效的数据比较,生物学解释和假设生成
数据库的引用
- 使用 NetworkAnalyst数据库发表论文时请注意引用文献(PMID:30931480、PMID: 25950236、PMID: 24861621、PMID:24078684、PMID:23766290)以及所用数据源对应的文献
电脑和浏览器要求
- 电脑:至少4GB内存
- 浏览器:启用Java标本的Web浏览器
- Chrome
- Safari
- Firefox
- IE 9+
支持的数据类型
- 芯片和测序数据(50M限制)
- 数据需要格式化(详见FAQs部份)
分析内容
- 差异分析
- 功能分析
- 网络分析
新用户注册
- 注册用户可以保存其工作状态
- 最多可以保存10个项目,每个项目最长保存1年
Network Analyst主页面
- 在浏览器输入 www.networkanalyst.ca 以进入主页面
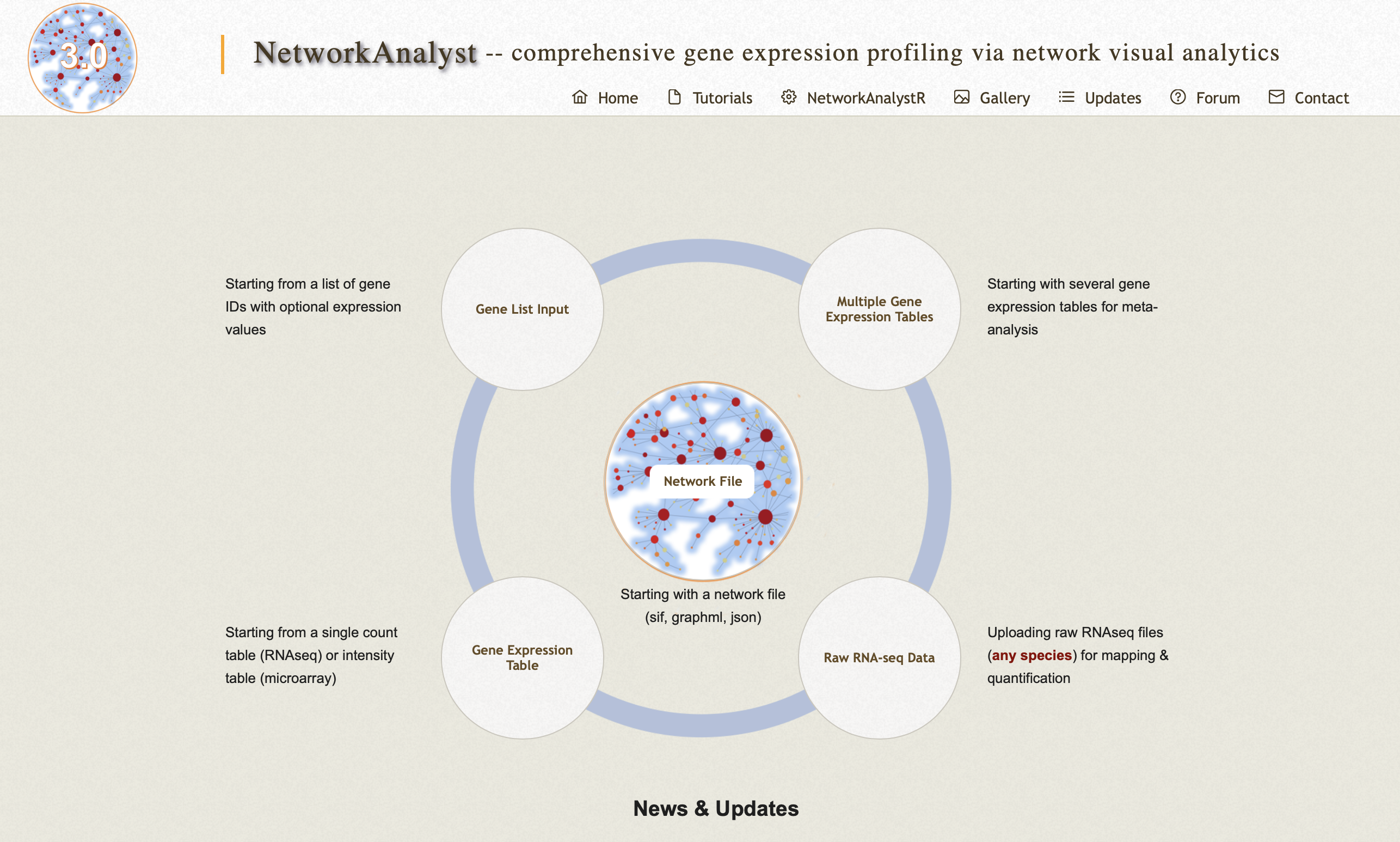
Gene List Input
- 基因列表
- 带有表达值的基因ID列表的差异分析
Gene Expression Table
- 基因表达数据
- 单个RNA测序或芯片表达数据的差异分析
Multiple Gene Expression Tables
- 多个基因表达数据
- 多个RNA测序或基因表达数据进行meta-analysis
Raw RNA-seq Data
- RNA原始测序数据
- 将RNAseq fastq文件上传到Galaxy服务器,以进行分析
Network File
- 网络文件
- 支持sif、graphml、json格式的网络分析
基因表达分析
- 这个部份的教程主要针对Gene Expression Table模块展开
- 点开以后的主界面长这样(可能会有一些跳转页面)
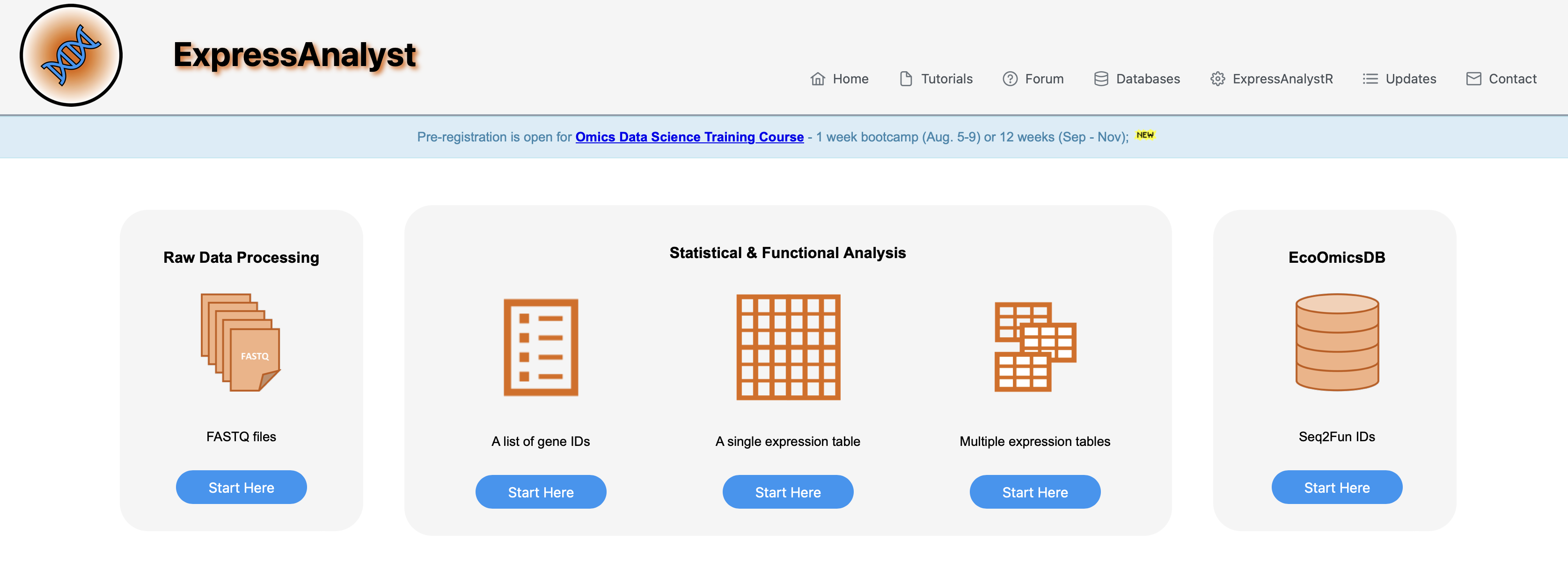
- 选择中间的”A single expression table”
上传数据
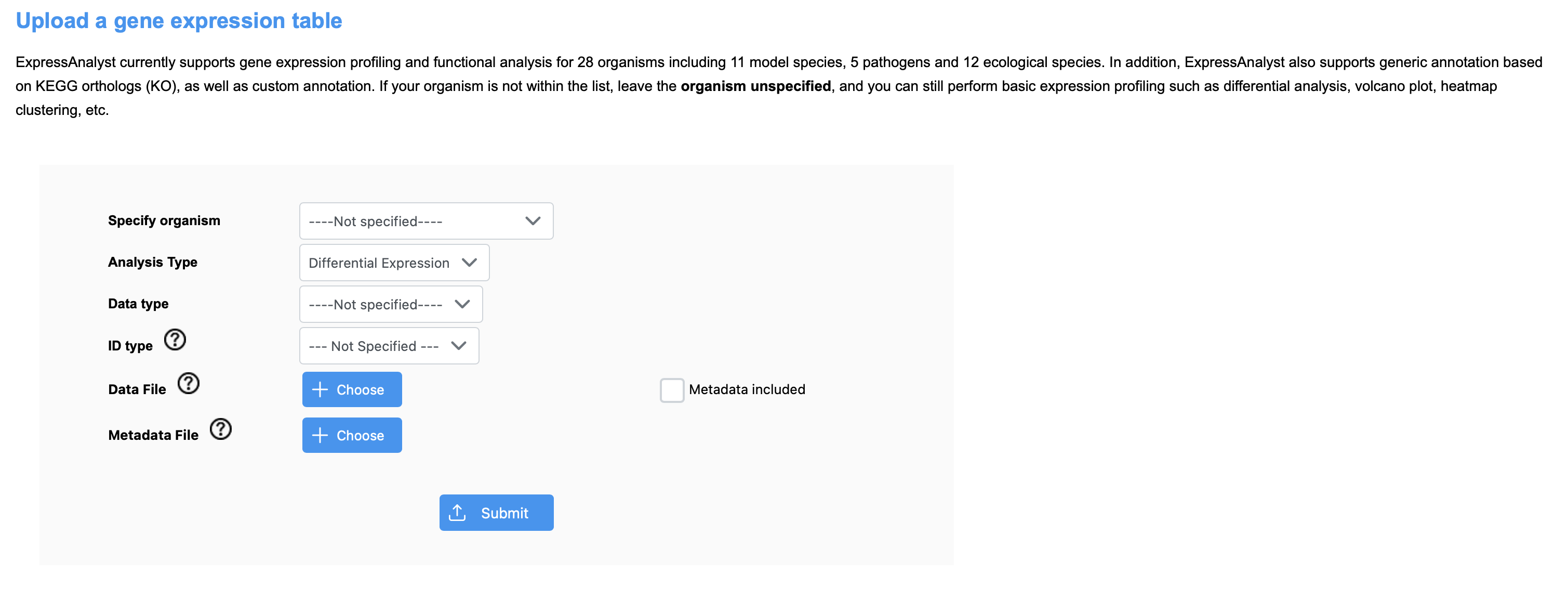
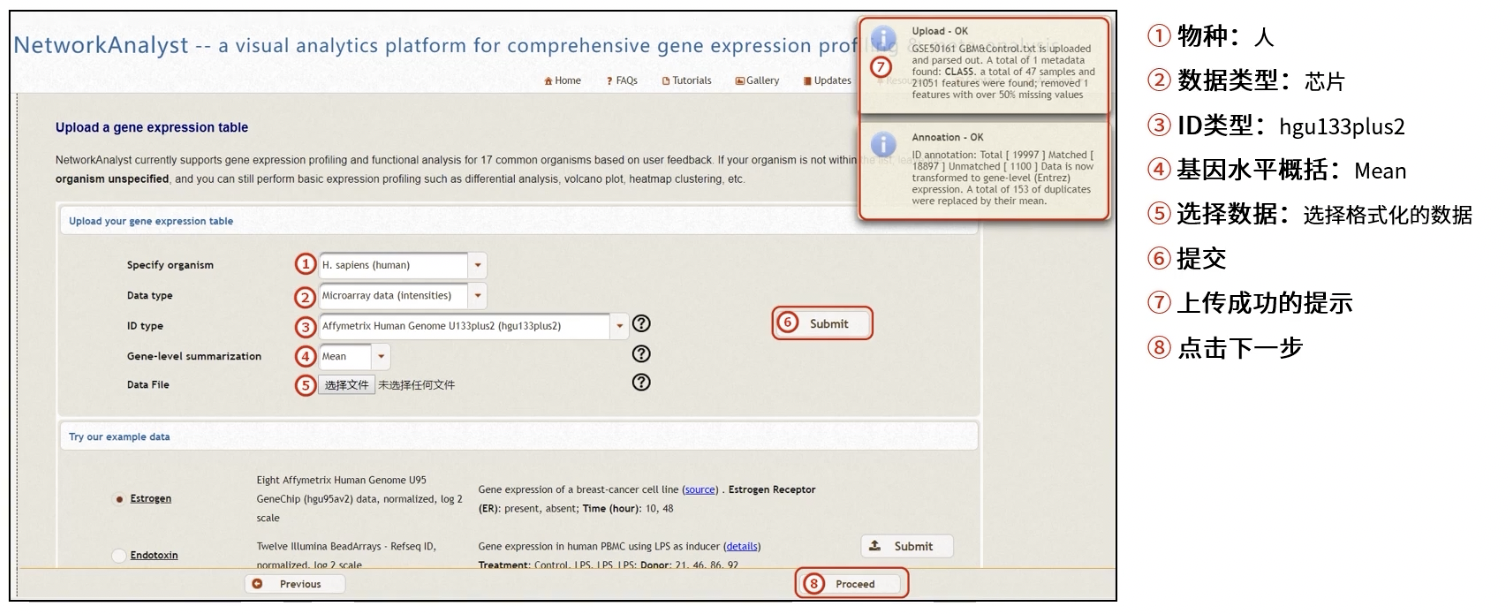
- 选择物种
- 选择分析类型(差异表达/时间序列或效应反应)
- 选择数据类型:芯片或测序
- 选择ID类型
- (好像没看到)选择基因水平总结方法:芯片选择Mean或Median,测序选择SUM
- 上传数据文件:50M限制
查看数据质检结果
以文字方式呈现:
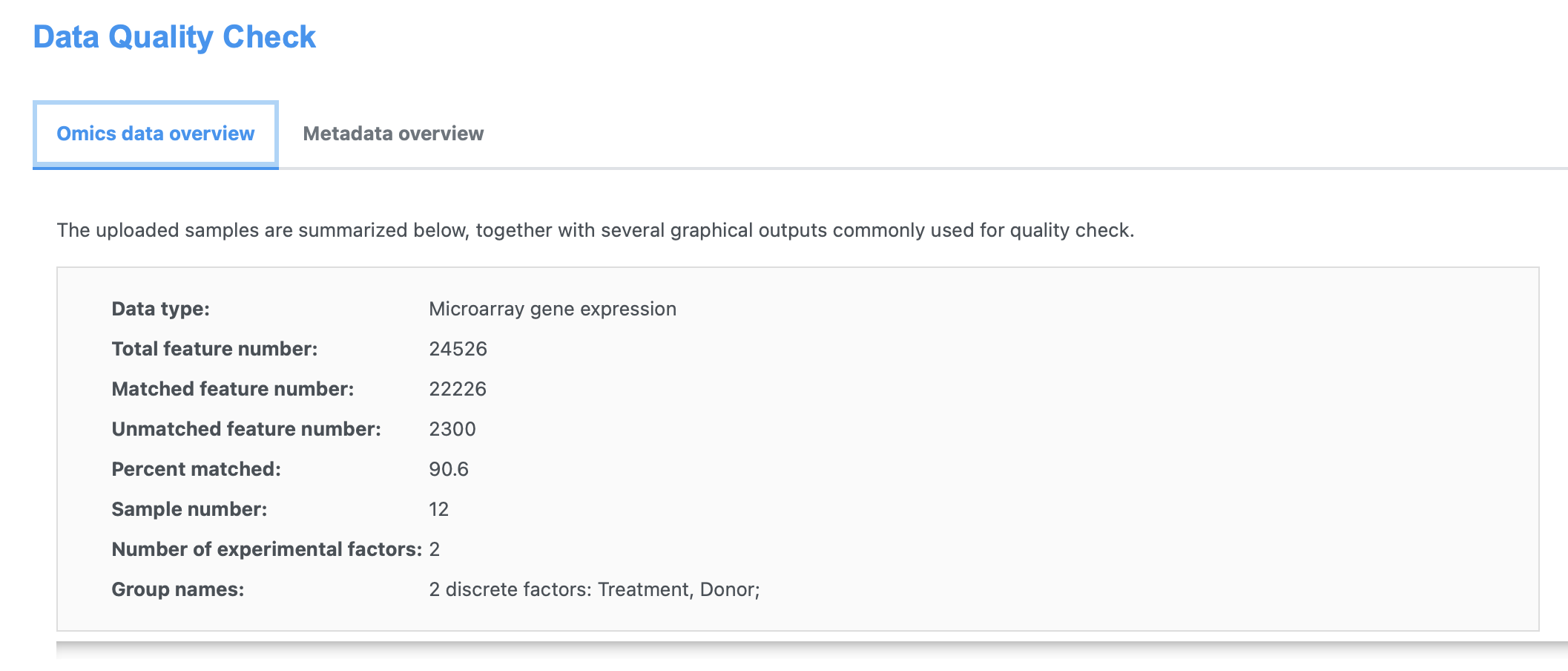
以图片方式呈现:
- 箱式图
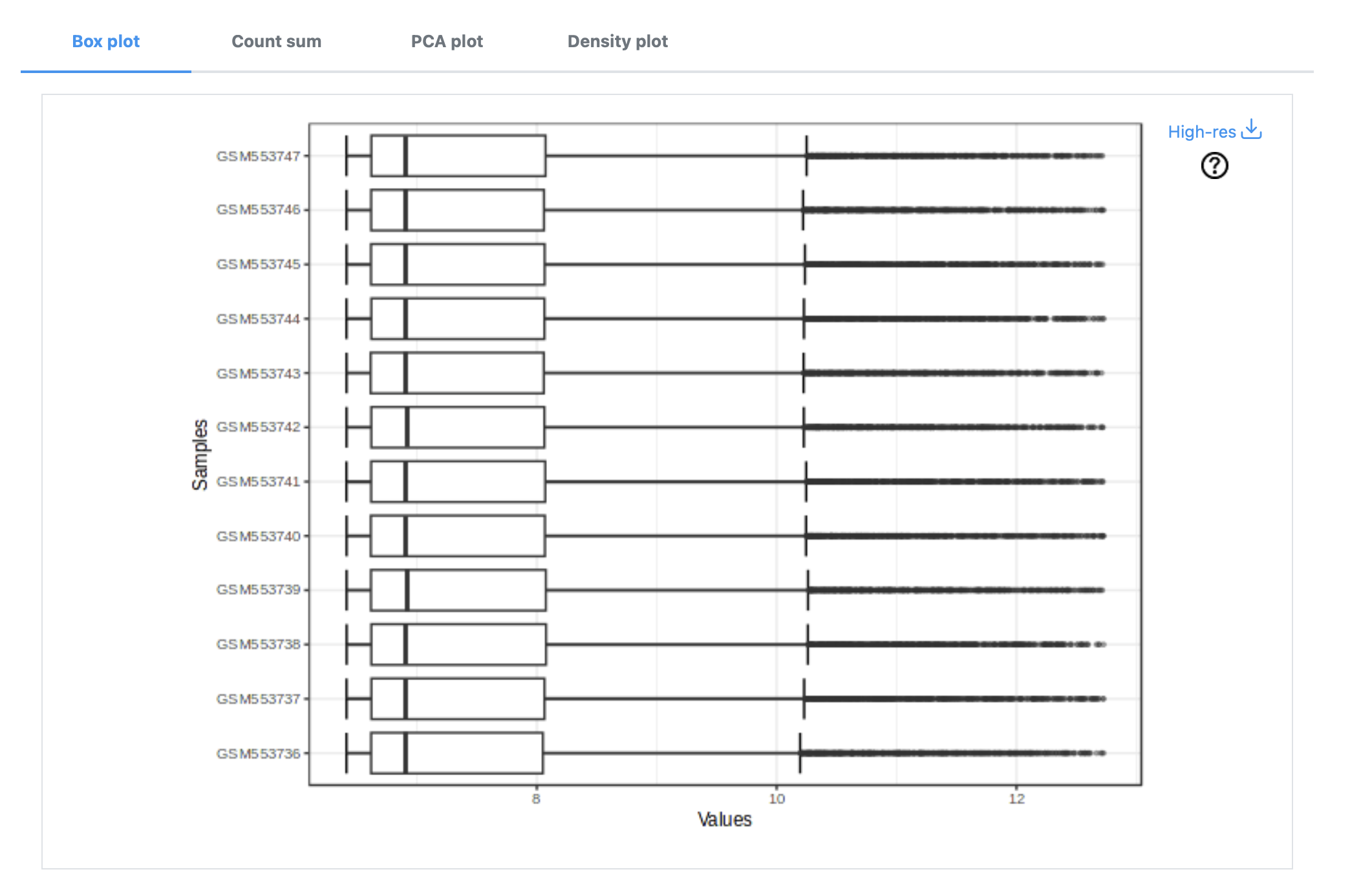
* 该组数据已经经过归一化处理,各分组具有相同的分布,并且所有数值都略大于8
* 箱式图可以用来判断数据有没有经过对数转换,芯片数据经过对数转换后,数据值通常<16,测序数据经过数据转换后数据通常<20
* 如果所有数据值的数据都<20,而且各组数据具有相似的分布,就可以合理地假设该数据已经经过了归一化 - 基因计数总和
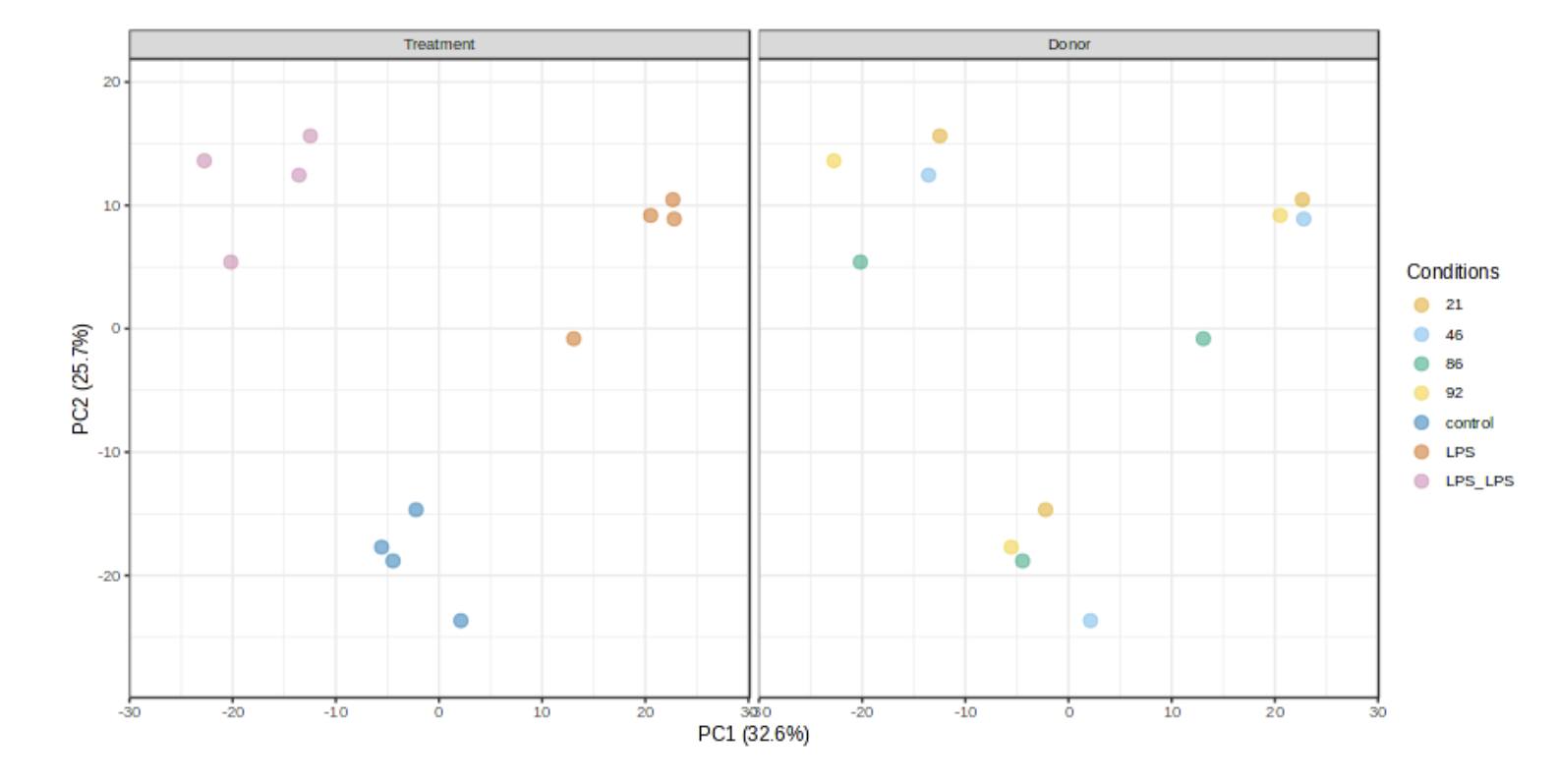
- PCA图:将高维数据投影到低维,来直观地识别表达模式
- 密度图
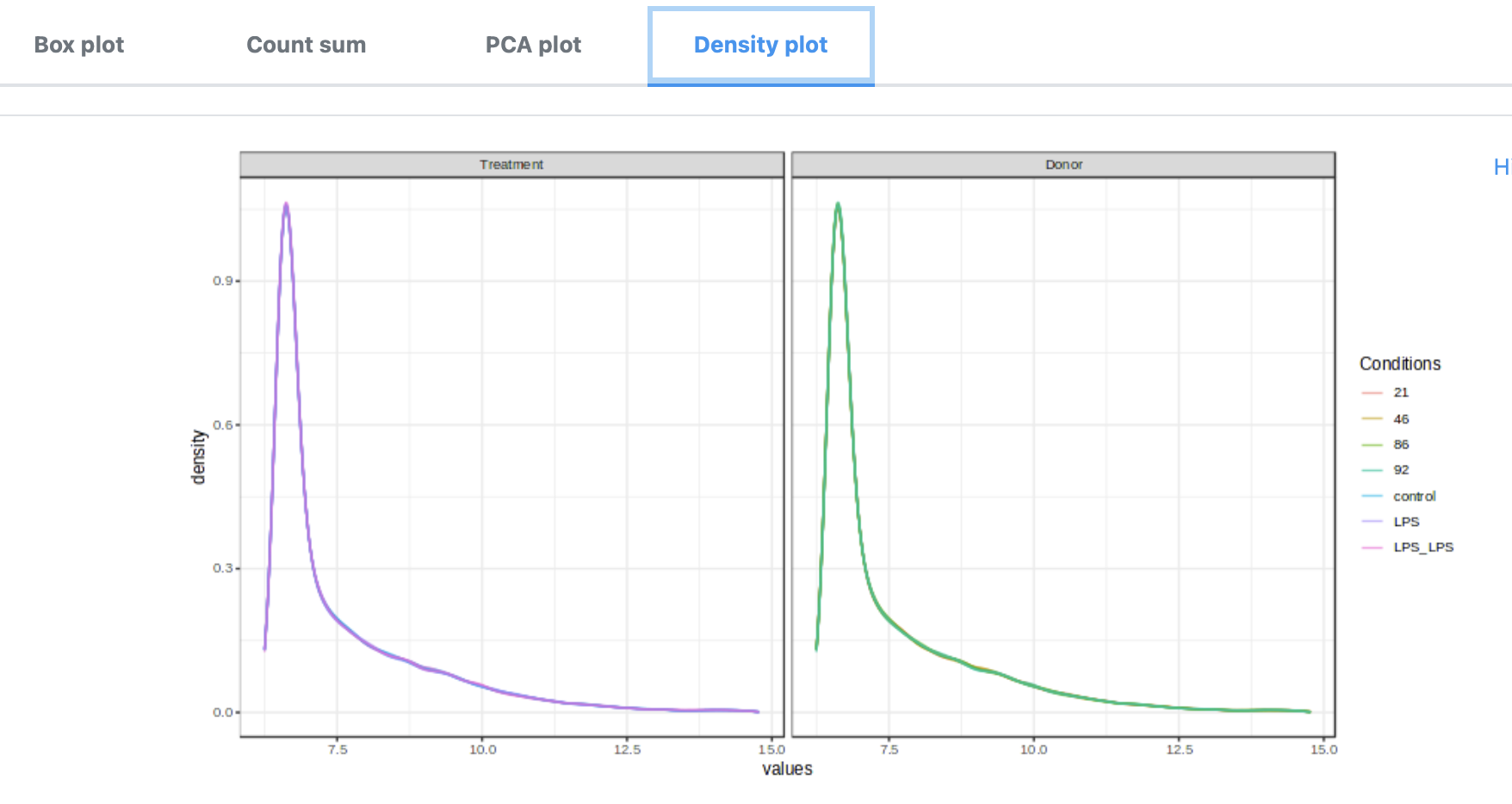
- 点击”Proceed”可以进入下一步
归一化
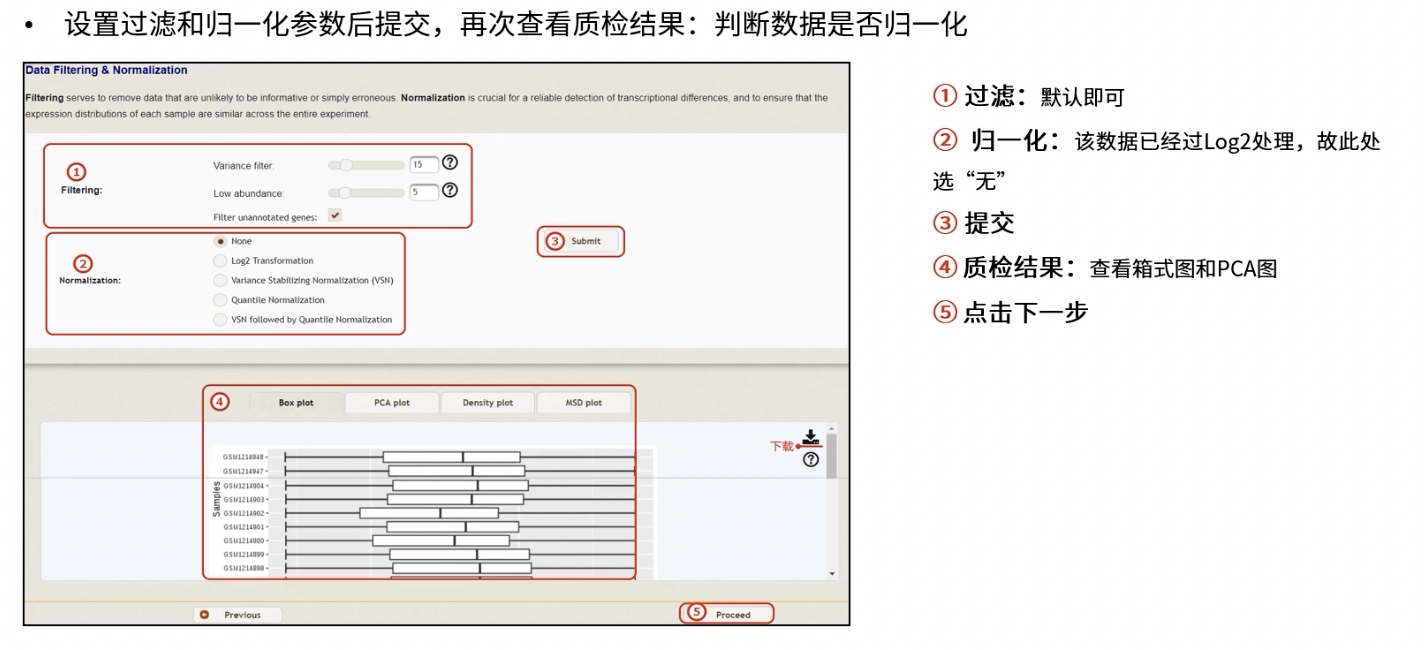
- 这一步的全程是数据过滤与归一化Data Filtering & Normalization
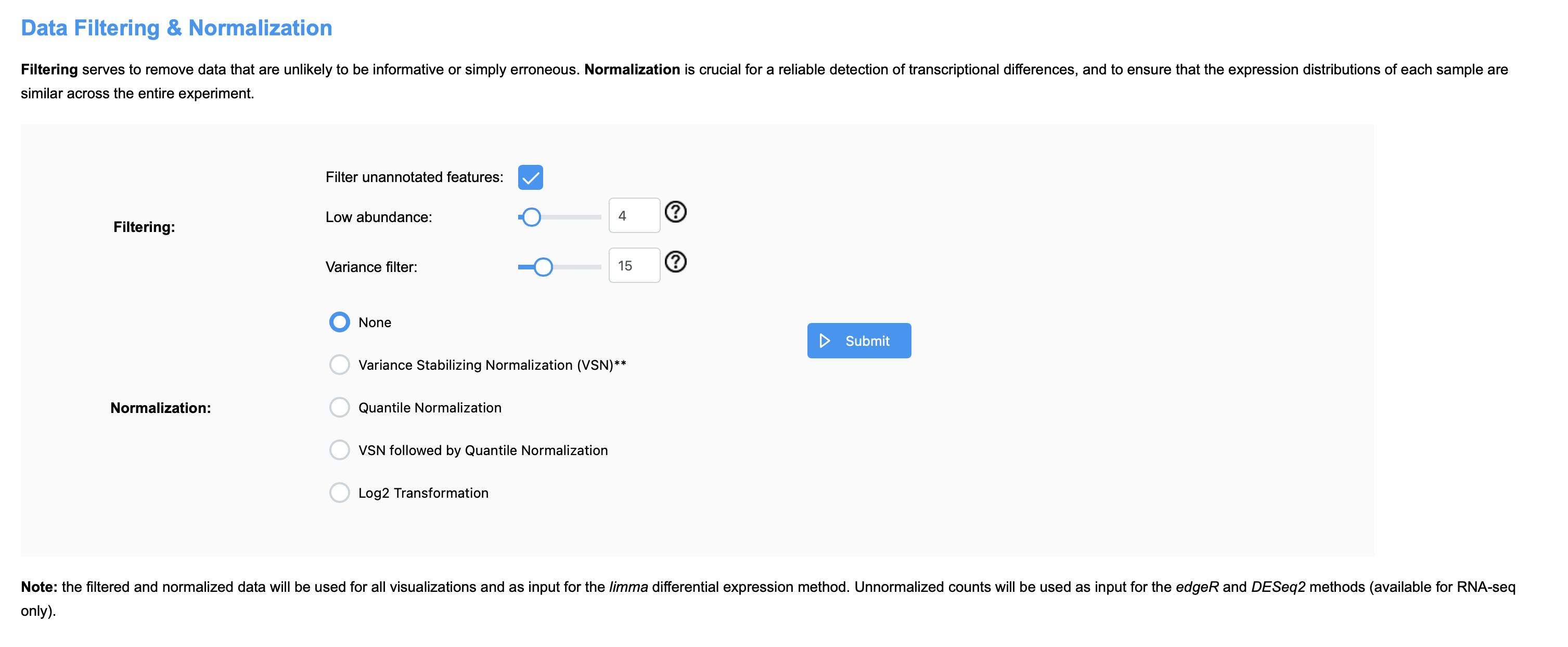
- 过滤这一步骤可以删除无用的数据,过滤的参数通常保持默认即可
- 归一化方式通常选择Log2 Transformation,这对于得出合理的结论至关重要(因为我们选择的示例数据已经经过了转换,所以此处选无)
- 点击提交,查看更新后的质检图
- 点击”Proceed”可以进入下一步
差异分析
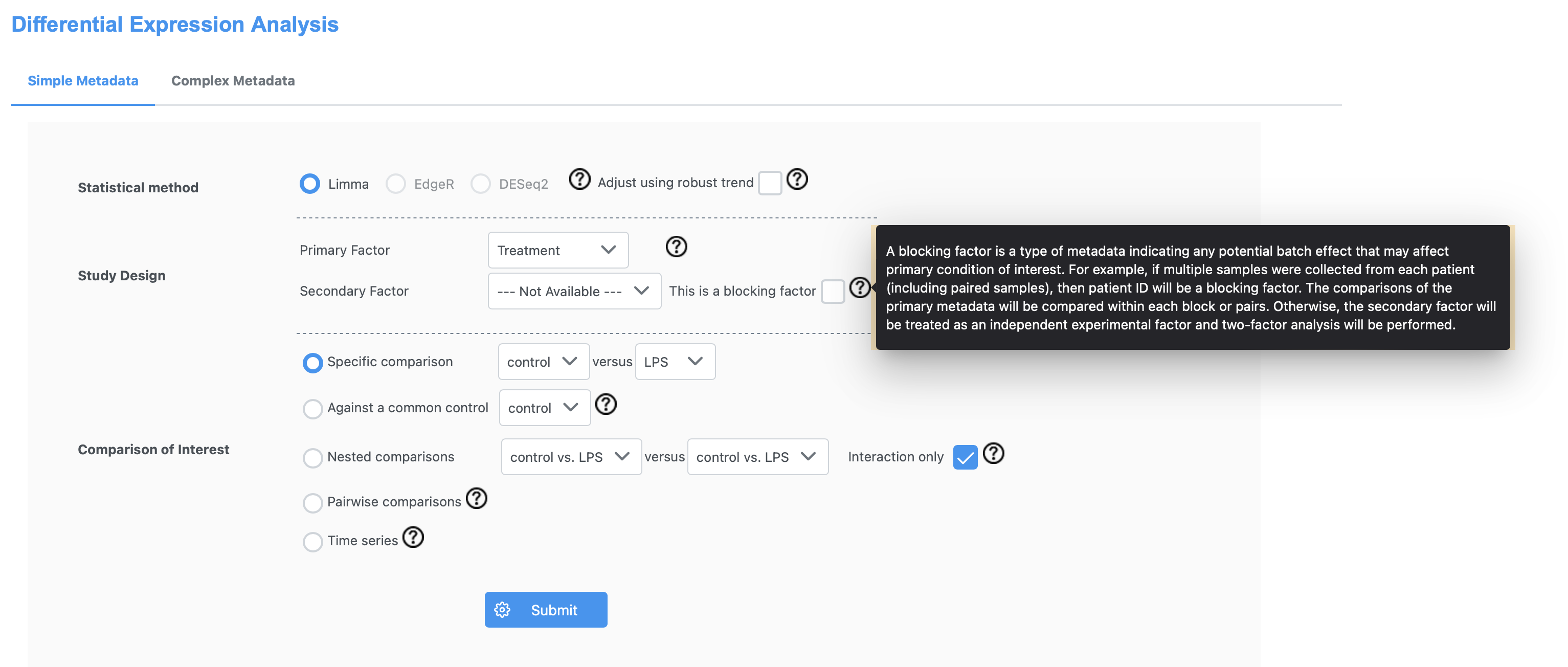
统计方法
- limma用于芯片分析
- EdgeR和DESeq2用于RNAseq数据分析
研究设计
- 主要因素
- 次要因素
比较方法
- 特定的两组比较
- 与一个共同的对照组比较
- 嵌套比较
- 两两比较
- 时间序列比较
- 不同比较方法的差异,详见FAQs部份
显著性阈值
- 调整后的p值,推荐0.05
- Log2差异倍数绝对值,推荐1.0
结果总结
- 符合筛选条件的显著差异基因数量

差异基因
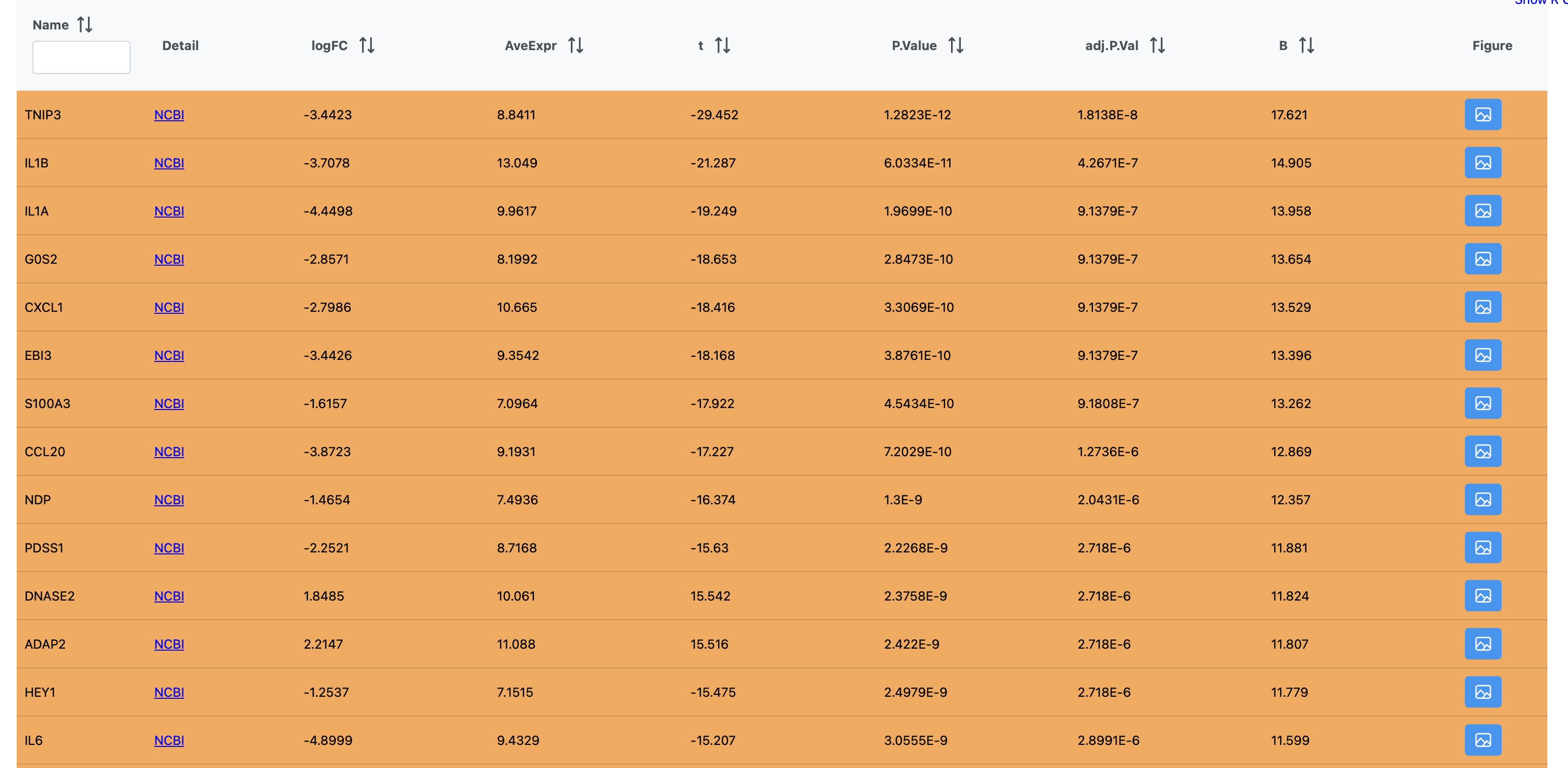
- 符合筛选条件的显著差异基因分析结果
- 排序依据:常用logFC或adj.P.Val
- 排序方式:升序或降序
- 点击Figure这一栏的图片,可以看到单个基因在不同分组中表达的箱式图
差异表达分析
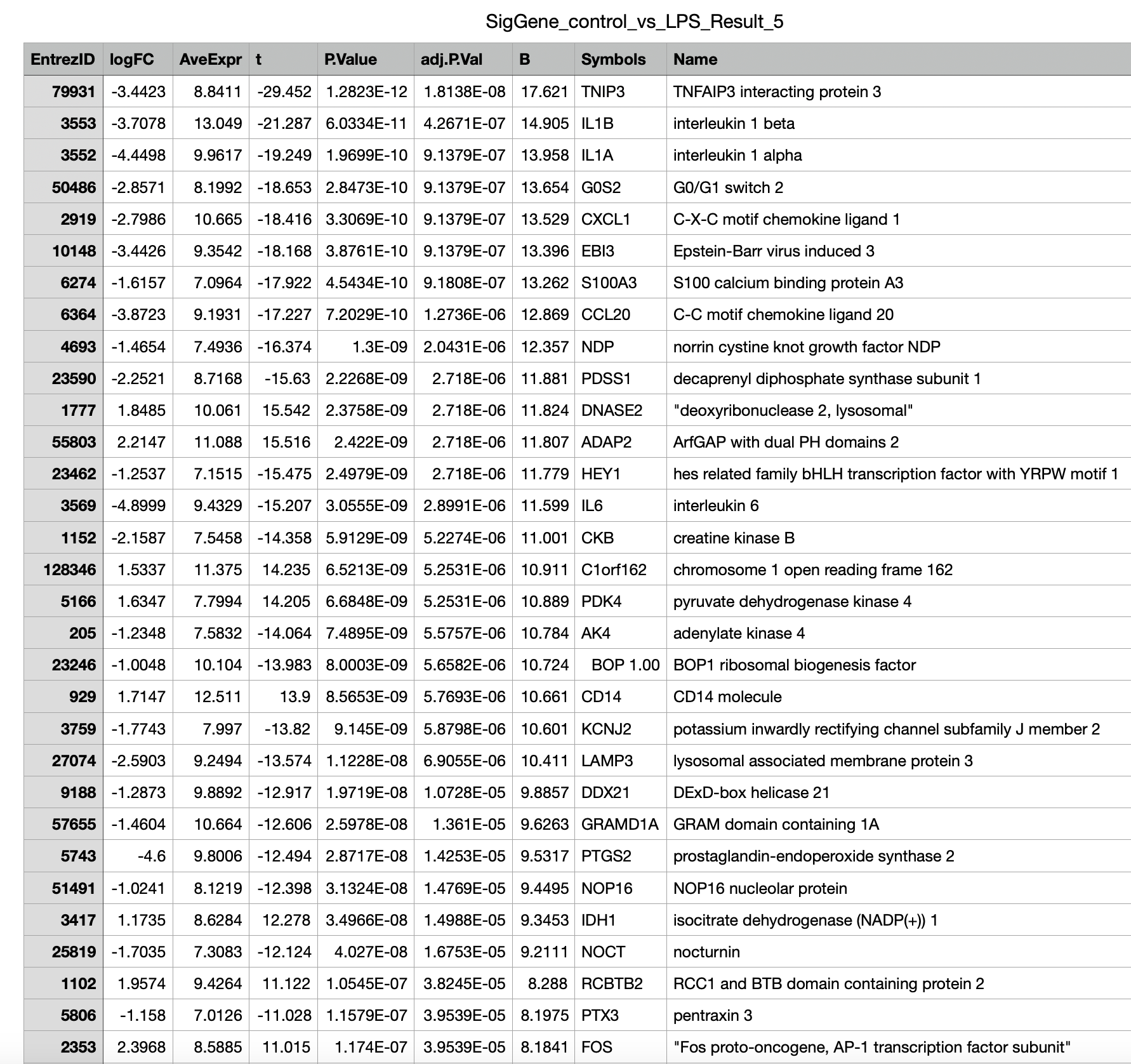
- EntrezID:NCBI Gene数据库基因编号
- logFC:log2 之后的fold-change
- AveExpr:标准化后基因平均表达值
- t:t检验
- P.Value: 原始P值
- adj.P.Val: 调整后的P值
- B: BH值
- Symbols: 基因符号
- Name:基因名称
应用示例
示例文章
- 示例文章:PMID:30356407
- 题目:利用WGCNA研究胶质母细胞瘤的候选生物标志物及其分子机制
- 杂志:BioMed Research Internation
- IF=2.2
芯片数据与筛选标准
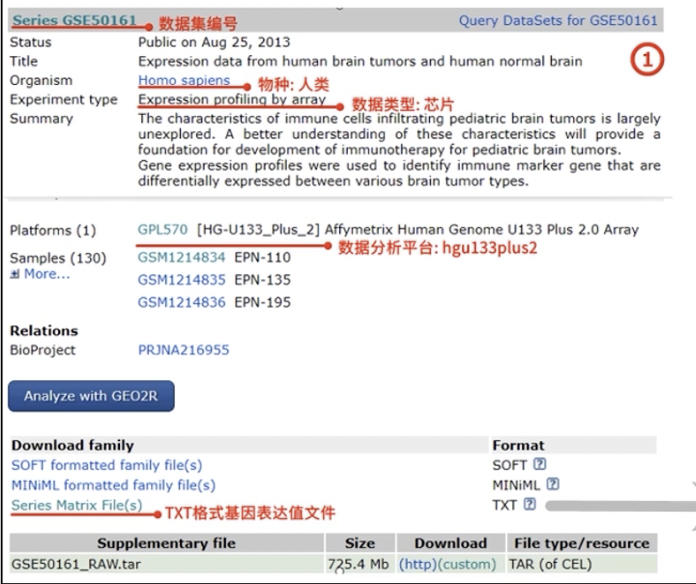
- 来源:GEO数据库:GSE50161
- 分组
- 实验组:34例胶质母细胞瘤患者的脑组织样本
- 对照组:13例癫痫儿童脑组织样本
- 差异分析
- 分析平台:R语言
- 筛选标准
- adj p value<0.05
- |log-fold change|>2
GEO数据下载
- 在GEO主页检索GSE50161,采集必要信息(如下图)后,下载.txt格式的基因表达值文件
格式化数据
格式化数据的要求
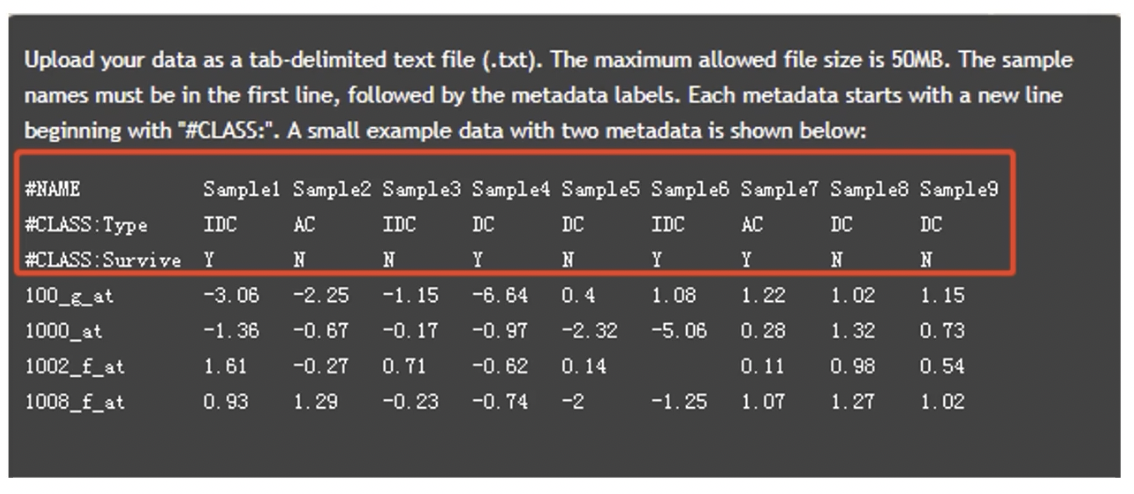
- 第一行:sample names样品名称
- 第二行:metadata labels 分组信息,需要以“#Class”开头
格式化数据需要进行的操作
- 把txt文件下载,用excel打开(numbers好像打不开这种类型的数据)
- 找到!Sample_title(对应样本标题)和!Sample_geo_accession(对应GEO数据库内的样本编号)
- 关注!Sample_title这一行的GBM样本,将所有样本为GBM的列复制到新的工作表中,把新的工作表重命名为sheet2
- 删除一些无用和空白部份,使表格变成下图的样子
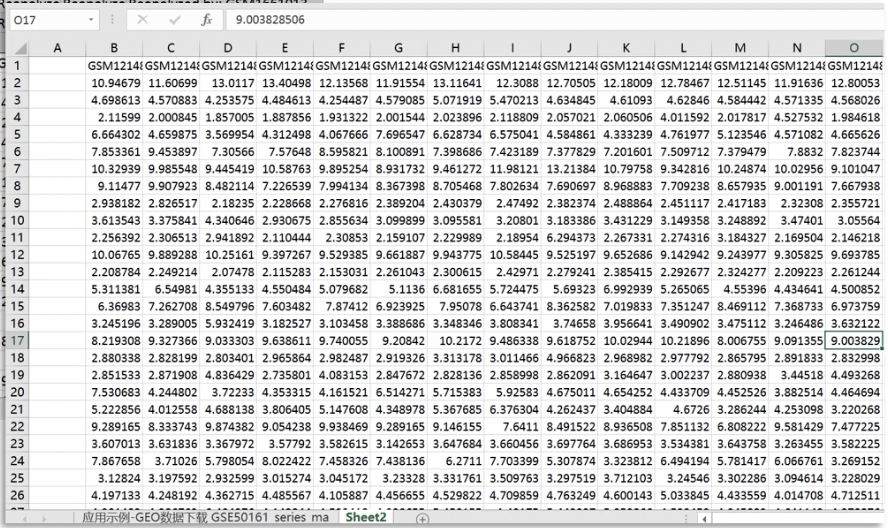
- 添加探针列,从原来的表格中复制到原文的正确位置
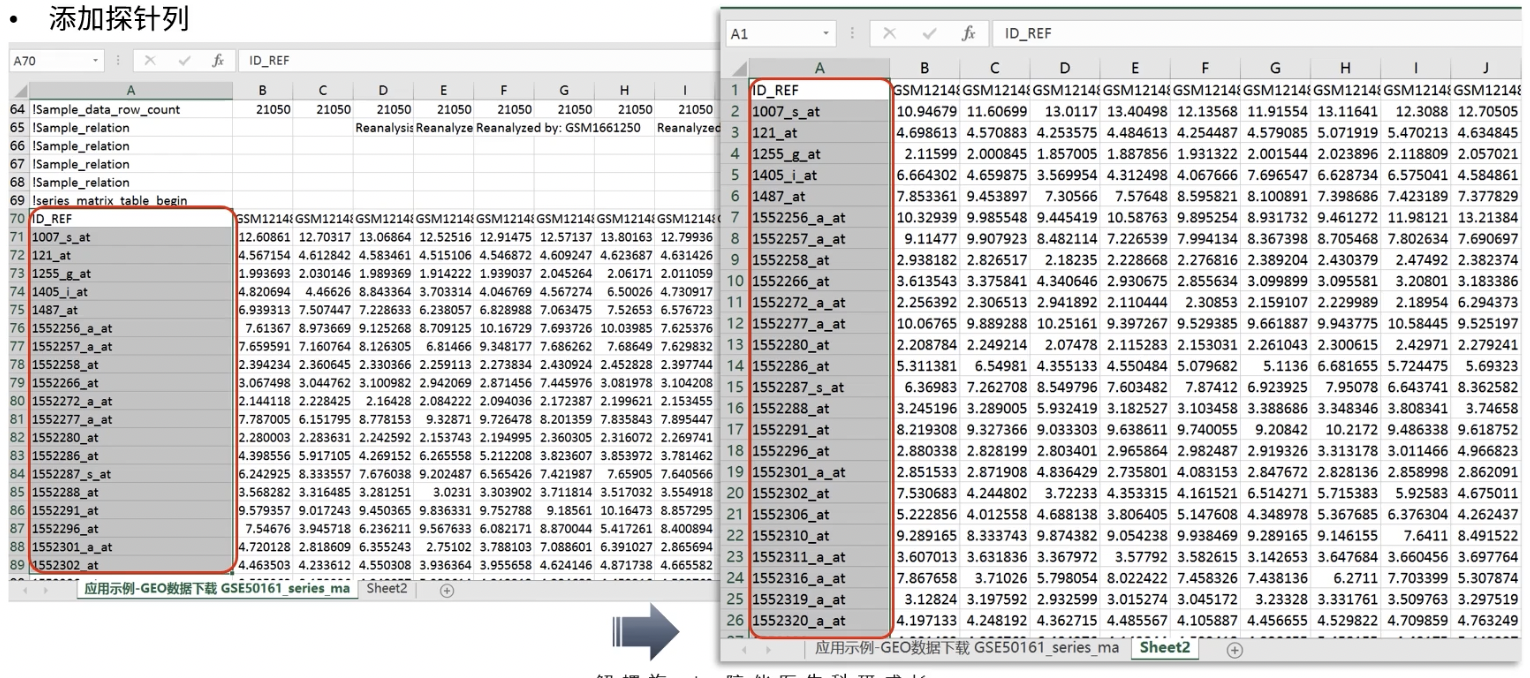
- 回顾NetworkAnalyst的格式要求,我们需要把”ID_REF”改为”#NAME”,在第一行后面插入一行,命名为”#CLASS”
- 最后,将sheet2整体复制,Ctrl/Cmd+A,新建一个表格并且粘贴,再另存为“文本文件(制表符分隔)”
上传数据
对ID type进行合适的调整
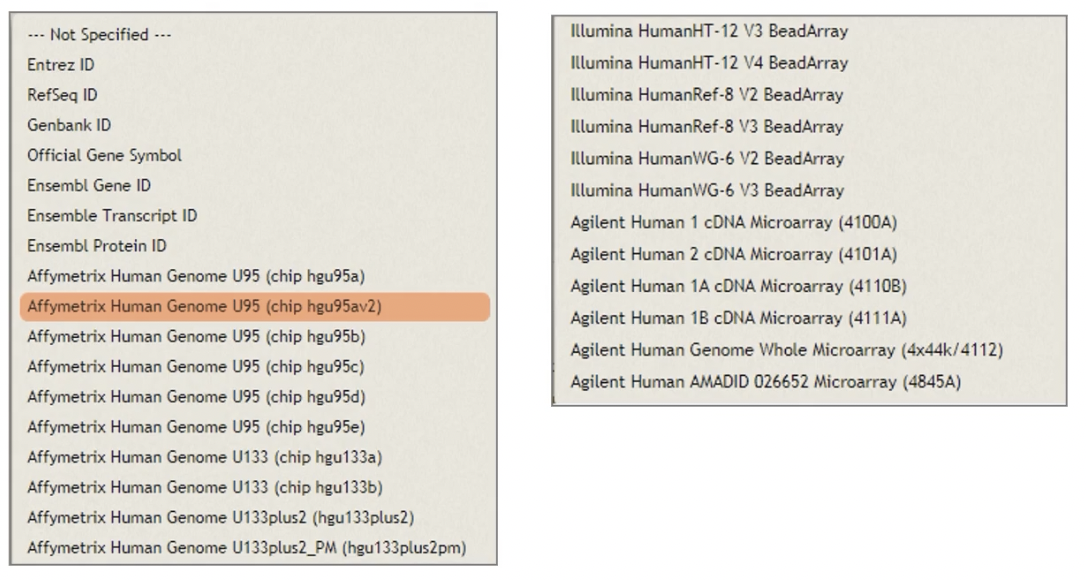
- Entrez ID/Gene ID,是一串数字,相对稳定,与Gene Symbol一一对应,可以用来做GO以及KEGG富集分析,参考网址https://www.ncbi.nlm.nih.gov/gene
- RefSeq ID:两个大些字母,加下划线,后面接数字,参考网址https://www.ncbi.nlm.nih.gov/gene/?term=2
- NC__:DNA序列
- NM__:mRNA序列
- NP__:蛋白质序列
- GenBank ID:全称是Gene Bank Accession Number,目前基本上与RefSeq ID一致
- Official Gene Symbol:HGNC数据库为基因提供的官方命名,由大写字母和数字组成,官方链接https://www.genenames.org
- Ensembl Gene ID:物种前缀+序列类型+数字
- 前缀ENS——Homo sapiens即人
- 前缀ENSMUS——Mus musculus即小鼠
- 序列类型G——Gene
- 序列类型T——Transcript转录本
- 序列类型P——Protein
质检
- 查看质检结果:数据概览和箱式图,PCA图等信息,判断样本的质量
过滤和归一化
差异分析
- 设置分析方法和分组比较信息后提交
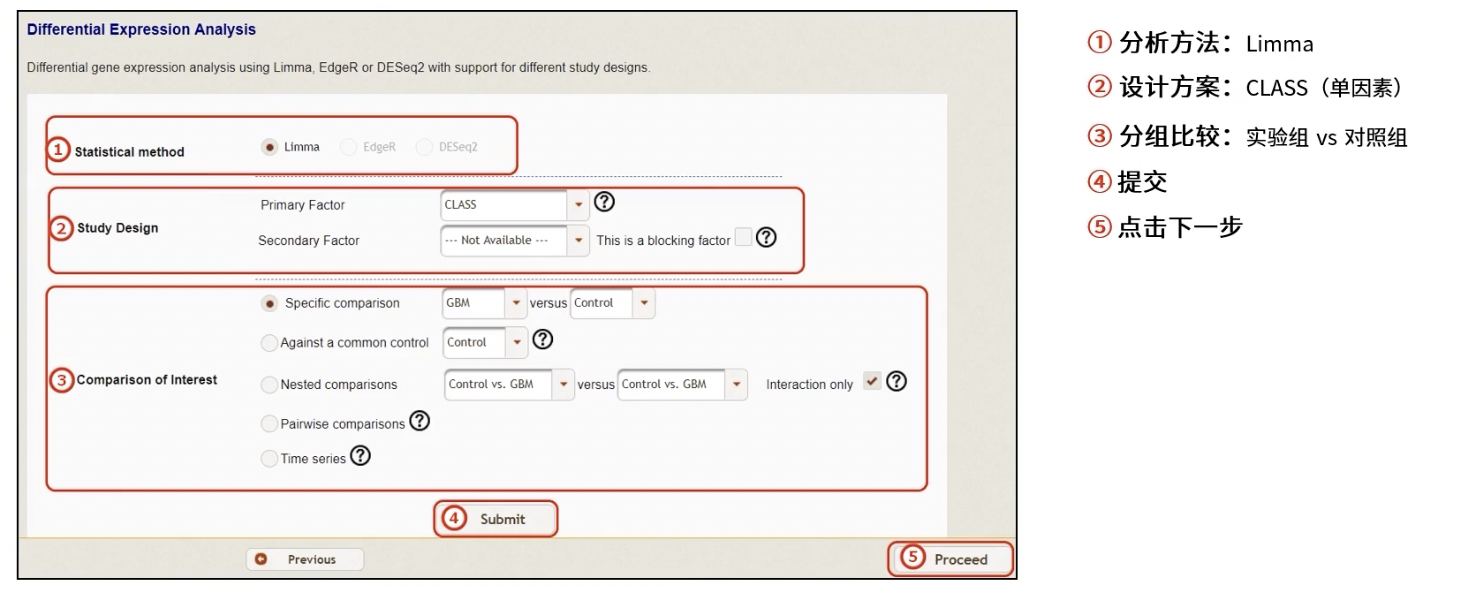
- 设定筛选标准后提交,查看结果概要并下载分析结果

芯片Meta Analysis
简介
什么是荟萃分析
- 荟萃分析是一种统计技术,用来整合已经在相似实验条件下收集的多个独立数据集,以获得更加强大的生物标记。通过组合多个数据集,该方法可以提交统计功效(更加多的样本)并减少潜在的偏差
- 通常不建议直接组合不同的独立数据集(即将这些数据集合并到一个大表格内),并将它们作为一个单元分析,这是由于与每个数据集相关联的潜在批次效应,可以完全压倒生物学效应
- 通常根据汇总统计信息(p-值,效应大小等)来计算荟萃分析和识别可靠的生物标记
哪些数据集适合进行荟萃分析
- 具有相同假设或者相同机理基础的数据集
- 只有两个分组(对照组/治疗组)的数据集
- 相同类型ID的数据集,即检测平台相同,或者同一厂商的同一系列芯片
- 所有数据在相同的比例或范围内的数据集
实战部份
- 点击进入ExpressAnalyst的Multiple expression tables分析模块
数据上传
- 样本数最多1000个
- 点击”upload”可以上传数据,一次只能上传一个数据集
质检
- 需要核对的数据集的内容
- 上传数据集的名称
- ID转换
- Meta数据质检
- 可视化
- 标准化
- 差异表达分析
- 数据总结
- 是否选择该数据集
- 质检结果
- 数据类型
- 匹配的特征数
- 样本总数
- 分组名称
- 数据集名称
- 消除批次效应:要记得勾选”Combat”后,点击”Update”
- 质检图
- PCA图
- 密度图
荟萃分析
- 有以下四种方法可以选择,选择合适的meta分析方法后点”Submit”,提交成功后点”Proceed”
- P值法
- 选择统计分析方法
- Stouffer:基于样本大小,仅用在所有研究质量相似的时候
- Fisher:无权重方法,比较推荐
- 设置p值:默认0.05
- 点击”Submit”
- 选择统计分析方法
- 效果大小法
- 定义:两组均值之差除以标准差
- Cochran Q检验:当数据与基线重合时,选择FEM,否则选择REM
- 统计方法:
- 固定效应模型FEM
- 随机效应模型REM
- p值:默认0.05
- 选择合适的统计方法后点”Submit”
- 投票计数法
- 根据阈值(p值和计数次数)选择差异表达的基因
- 不推荐使用
- 直接合并法
- 将所有数据集直接合并成一个大型数据集,然后对合并后的单个数据集进行分析
- 不推荐使用
- 选择合适的meta方法后,点击Proceed
Meta分析
- 参数设置
- 单个数据集的显示方式
- 排序依据
- 排序方式
- 更新
- 参数设置
- 结果预览
- ID类型
- 单个数据集差异倍数
- 整合的差异倍数
- p值
- 箱式图预览
- 点击Proceed
FAQs
如果我的芯片检测平台不在支持的列表中,怎么办
- 芯片官网或GEO数据库下载芯片注释文件
- 使用芯片注释文件为探针注释常见的基因ID(如Official Gene Symbol, Entrez, Refseq, Ensemble等)
- NetworkAnalyst上传数据时:ID类型选择:未指定”,然后上传已经注释的数据文件
如何格式化分析数据
- 文件格式:制表符分隔的文本文件
- 数据格式(示例:2个实验条件以#+大写字母的形式展示出来,参考下图)
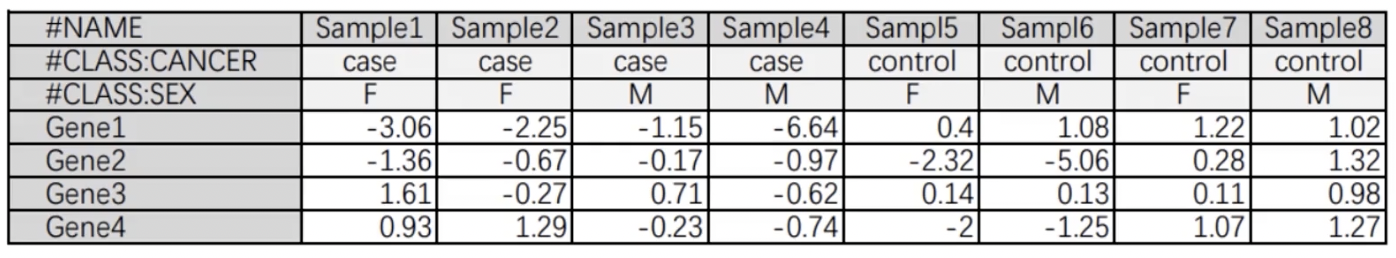
如何判断数据是否归一化,并选择合适的归一化方法?
- 箱式图可以判断数据是否经过了对数转换,芯片数据对数转换后,数据值通常<16,测序数据对数转换后数据值通常<20
- 如果所有数据值都<20,并且各个分组都有相似的分布,就可以合理地解释该数据已经归一化了
- NetworkAnalyst所有归一化方法都在以前的研究中使用过,它们基于稍有不同的假设,并且产生基本相似的结果
如何识别并且处理质检一场的样本
- PCA图可以识别潜在的异常样本,这些异常样本通常是远离其余样本的离群值
- 发现异常样本:
- 首先检查样本是否正确地被测量,在许多情况下,异常值时分析过程中操作错误的结果
- 如果无法校正这些值,则应删除样品,然后重新开始分析
如何选择合适的差异分析方法
特定两组比较
- 适用于单因素实验设计
成对比较(两两比较)
- 适用于任何两两分组之间的比较
- 举例,取A、B、C三个组,“成对比较”将比较AB、AC和BC
时间序列比较
- 适用于连续分组(多个时间点或不同浓度)的比较
- 举例,取A、B、C三个时间点,“时间序列比较”将对比AB和BC
嵌套比较
- 适用于因条件不同,而产生不同反应的基因
- 举例,在10小时和48小时,测量带有和不带有雌激素受体细胞的差异表达基因