GEO2R基本介绍
- GEO2R 是GEO数据库提供的在线工具。它提供了一个简单的页面,允许用户对复杂的数据进行分析,主要用于比较2组或多组样本,以获得差异性表达的基因。
- GEO2R工具后端使用已建立的bioconductor r包来转换和分析复杂的GEO数据,并将结果显示为按重要性排序的基因表,可以通过表达谱图可视化。
- GEO 2R主要是对系列数据(series)进行分析,但不是所有系列数据都能用GEO2R工具进行分析,比如大多数测序数据等,不能使用GEO2R,对于这类数据,工具栏中的 “Analyze with GEO2R” 按钮不会显示。
- 使用GEO数据库发表文献请注意引用GEO的文献(PMID:11752295、PMID: 23193258)以及所用数据源对应的文献
GEO2R操作简介
数据检索
- 在浏览器中输入:https://www.ncbi.nlm.nih.gov/gds
- 或在NCBI主页进入GEO DataSets,检索栏里输入关键词,点击“search”按钮
- 在显示的页面中的筛选条件“study type”中找到expression profiling by array
- 在系列数据页面(其实从文章里点进去也行),点击“Analyze with GEO2R“的按钮,进入工具页面
工具页面
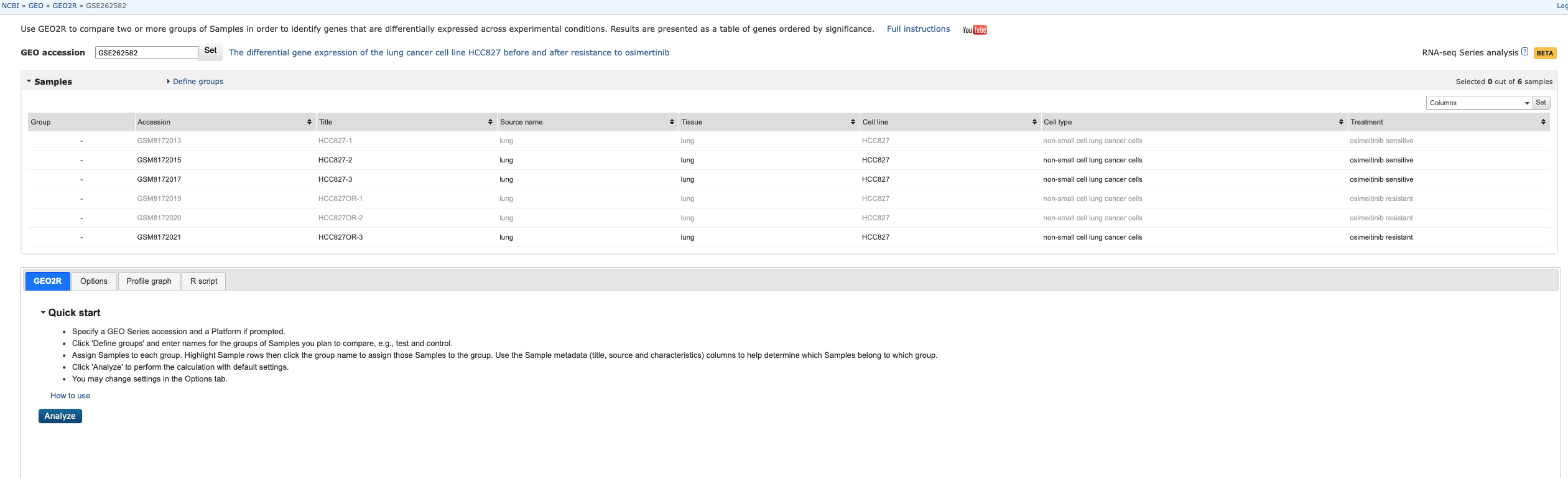
- 最上面的”Use GEO2R to compare two or more…”是对GEO2R功能的简单描述,点击”Full instruction”可以查看详细的使用说明,旁边的”YouTube”可以链接到Youtube查看视频说明
- 以”Geo accession”开头的:是当前数据集的ID,输入其他的ID点击Set即可更换
- 以”Samples”开头的内容:系列数据的信息和分组设置区,从做到右的信息分别是:
- 分组信息(因为当前还没有分组,这里全都是”-“)
- 样本的ID(好像被称为GSM ID)
- 样本名称
- 样本来源
- 细胞类型
- 组织类型
- 蓝色的“GEO2R”区域:是GEO2R分析工具区
- GEO2R:可以用GEO2R差异表达谱分析
- Value distribution(这里没看到):查看各组数据分布(通常用来看一下数据是否中位数中心化,即中位数位于同一水平线上,如果是,则表示数据已经经过了标准化,可以用于组间比较)
- Options:GEO2R分析参数调整,一般无需调整
- Profile graph:通过输入平台的检测单元ID,查看该检测单元的表达谱
- R script:显示当前计算机的R语言脚本
设置分组
- 本例中为研究奥希替尼敏感组与奥希替尼抵抗组的基因表达差异,因此可以分为sensitive组和resist组
- 当设置2个分组时,分析的是这两个分组的基因表达差异
- 设置2个组别时,计算的是组别2(下面的组)/组别1(上面的组)
- 点击“Define Groups“,在弹出的下拉框内填写第一个组别名称,按回车(图中的sensitive)
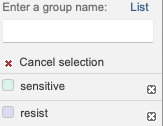
- 继续输入第二个组别的名称,按回车键(图中的resist)
- 选中sensitive样本(图中浅绿色),然后点击“sensitive”组,把样本和组别关联起来
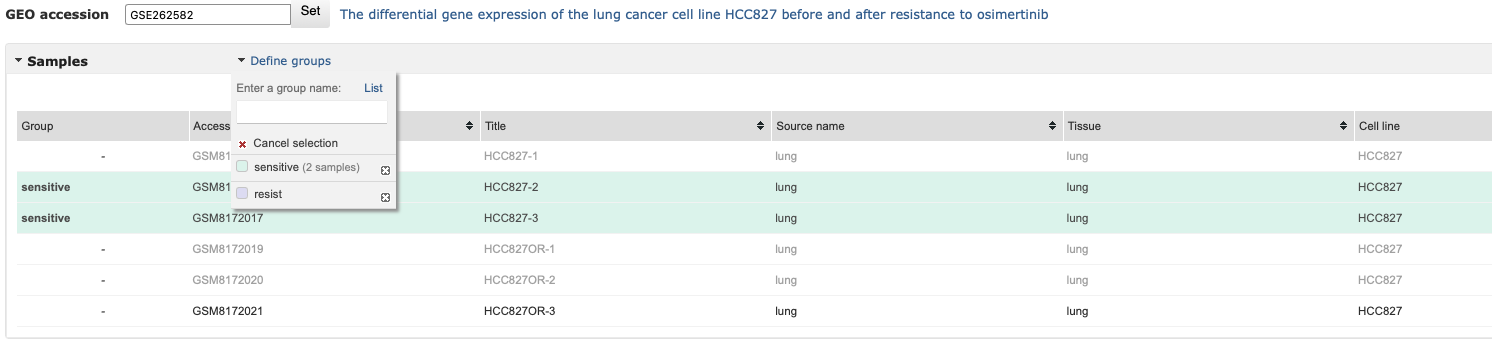
- 同理,选中resist样本(图中浅紫色),然后点击“resist”组,把样本和组别关联起来
- 点击“Analyze”按钮,查看分析结果,结果以表格显示,按照p-value排序,p-value越小,就越显著
对分析结果页面进行介绍
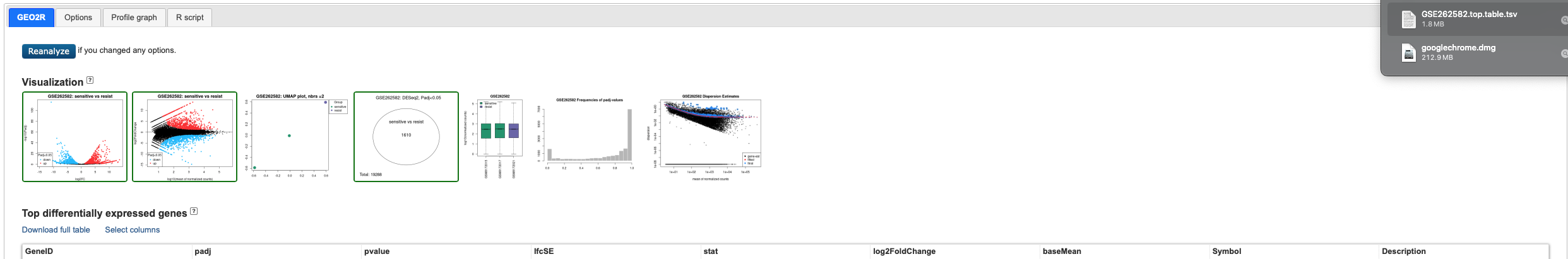
- GeneID:芯片的探针
- padj:调整后的p值(调整后的p值<0.05时就可以认为表达有差异了,在“options”一栏可以选择p值调整的统计学方法,默认为Benhamini&Hochberg(False discovery rate),一般选择默认就可以了)
- pvalue:即p值
- lfcSE:是指差异表达分析中的对数折叠变化标准误差(Log Fold Change Standard Error)。在基因表达分析中,通常使用对数折叠变化(Log Fold Change,LFC)来衡量基因在不同条件下的表达水平变化。而lfcSE则是对这个变化的标准误差的估计。简单来说,它表示了差异表达的可信度,越小则表明差异越显著可靠。
- stat:指统计学上的检验统计量(Statistic)。在差异表达分析中,这个统计量通常用于衡量两组样本之间基因表达水平的差异是否显著。常见的统计量包括 t 统计量、z 统计量等,它们的数值大小和显著性水平可以帮助确定基因是否在不同条件下呈现出显著的表达差异。
- log2FoldChange:倍比变化以2为底的对数值,负数为组别2低表达,正数为组别2高表达(是组别2/组别1的值再取2为底的对数得到的结果)
- baseMean: 一栏通常指基础均数(Base Mean)。在差异表达分析中,它表示基因在所有样本中的平均表达水平。换句话说,baseMean 是指所有样本中基因的表达水平的平均值,不考虑分组或条件。这个值可以帮助了解基因在研究中的整体表达水平,以及在后续分析中进行归一化和比较时的参考基线
- Symbol:这一栏通常指基因的符号(Symbol)。基因符号是基因的简称或缩写,用来标识和表示基因。它通常是基因名字的一种简化形式,便于在数据分析和报告中使用。基因符号通常是唯一的,它们用来代表特定的基因,使得基因在不同研究中更易于识别和比较。
- Description略
分析页面的结论
- 通常看一下LogFC和p值就可以,前者表示差异的幅度(绝对值越大表示差异幅度越大),后者指示是否具有统计学意义
- 关于调整后的p值和原始p值,调整后的p值是对p值再进行统计学分析,他们的统计结果未必一致,比较准确的是使用调整后P值,但文献中使用p值和调整后p值的都有,当调整后p值筛选后基因数过少时,可以使用p值
- 对于基因的注释,小伙伴们可关注生信体系课下篇内容
保存分析结果
GEO2R结果分析
- 目标是获取变化幅度大,且有统计学意义的基因群
- 在筛选前,要先求一下logFC的绝对值,方便后续的操作
筛选调整后p值<0.05的
筛选变化幅度≥2倍的
针对一个基因对应多个探针的情况,删除重复项
两种操作方法
- 保留logFc绝对值最大的,删除其余的
- 或者将所有logFc取均值
具体怎么做(保留最大的)
- 对绝对值那一行应用降序排序
- 然后把gene symbol中的重复项删掉
删除一个探针对应多个基因的项
删除无基因名称项
- 选中gene symbol,升序,最上面的项就是无基因名称的,可以被我们删去了
进一步处理分析结果
- 从差异基因中挑出一些作为目标基因,开始分子生物学套路
- 进行KEGG分析GEO分析和富集分析等
- 本例中我们是以2倍为标准线去划分“差异”基因的,这个标准可以适当调整,以获取更多或者更少的基因
- 如果遇到差异表达基因特别少的情况,除了本身两组间差异就不够显著以外,还可以考虑是由于原始数据的取样不够规范,导致不同组之间差异不大
GEO2R的多组比较
- 可以有,但不常见
- 返回的是任意两组有差异的基因
- 操作方法基本同前