


选择的文献

- Hallmark-特征
- 肿瘤学入门的必读文献,没有之一
关于作者的介绍

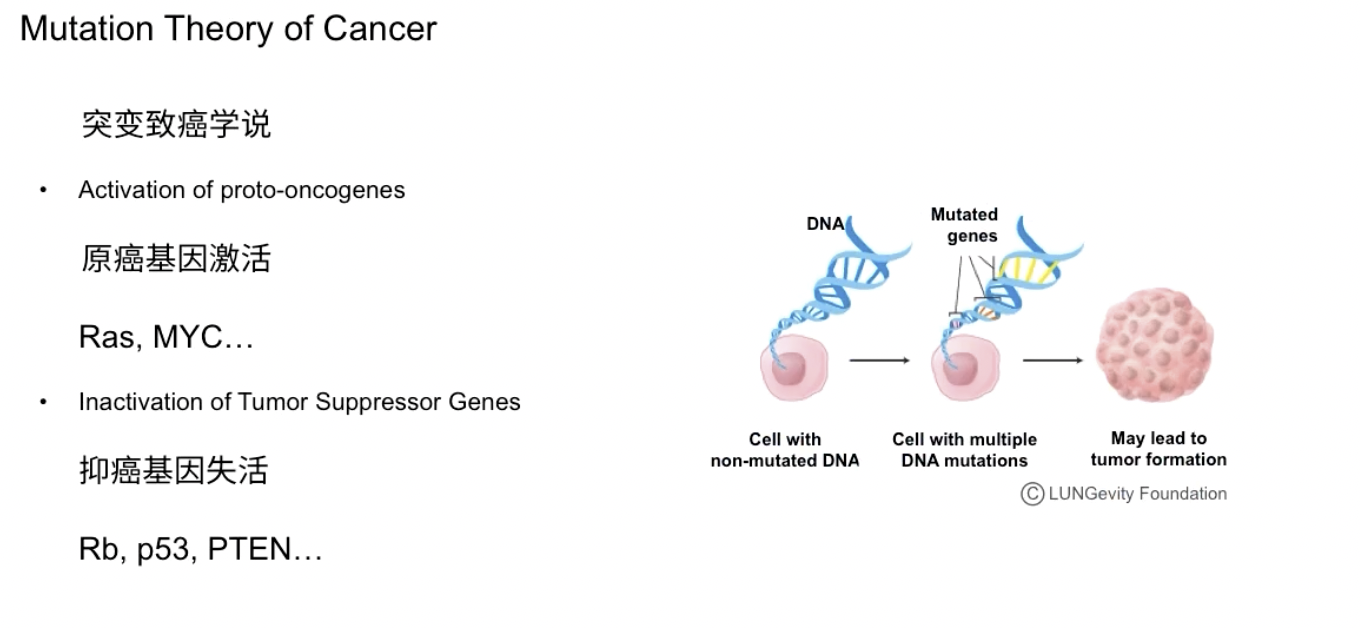
简单复习一下基因突变学说
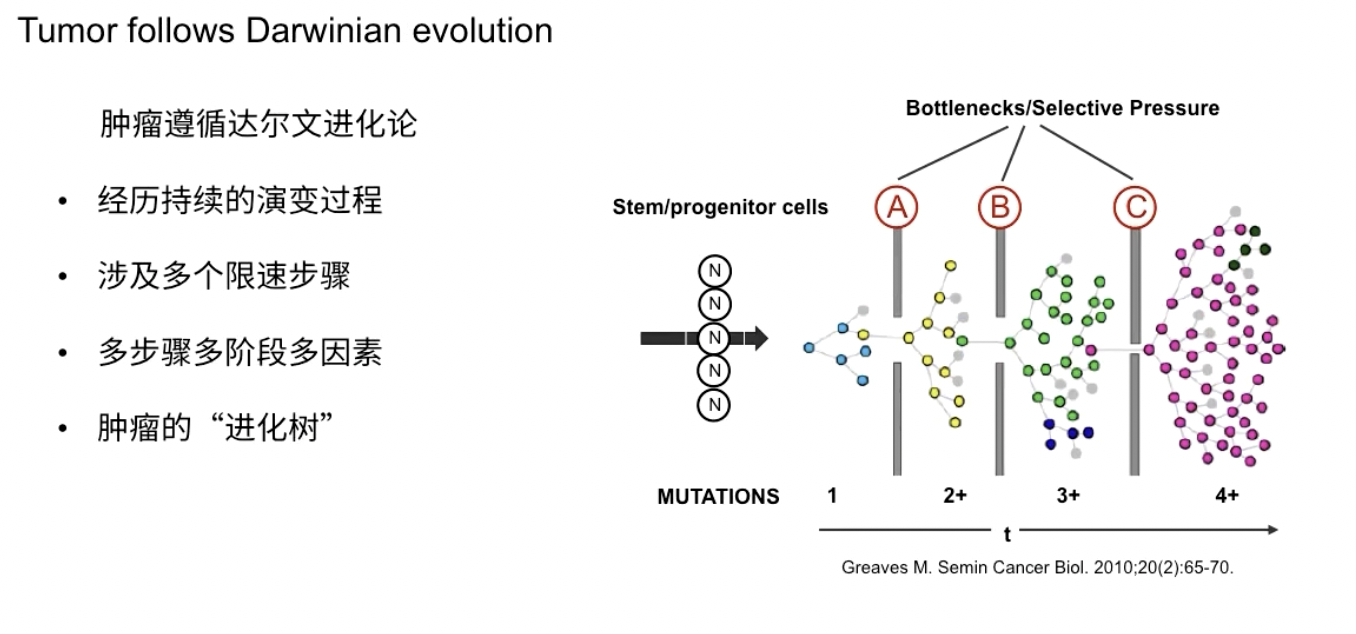
- 人类约30%的恶性肿瘤与原癌基因k-ras有关,其所在序列的某一位点突变可以活化ras基因,激活相关的信号通路,导致细胞的恶性增殖
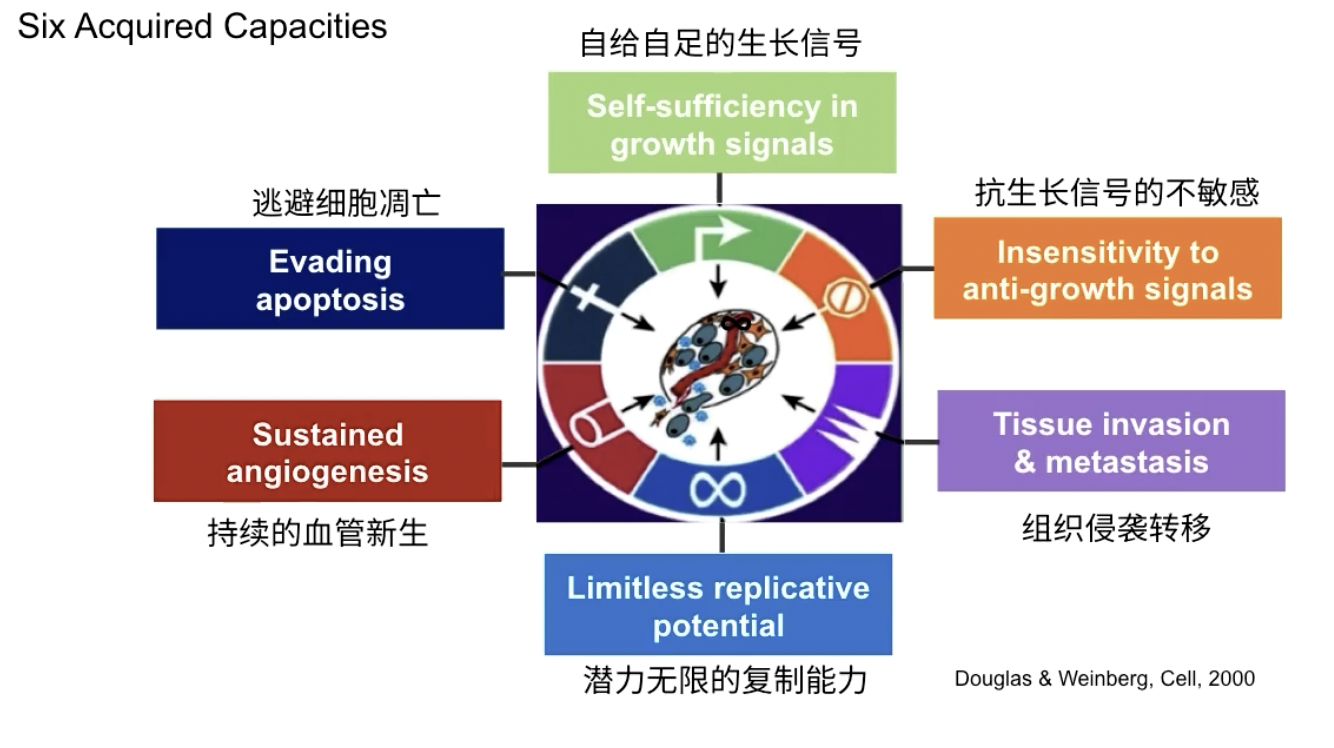
肿瘤的六大获得性特征
自给自足的生长信号
- 细胞生长的首要驱动因素是激活增殖的能力,就像田径比赛是裁判员发出的开始指令一样
- 很多癌基因都与细胞的生长调节密切相关,正常细胞需要生长信号的刺激才能进入有丝分裂,比如在体外培养细胞的过程中,正常细胞需要加入生长因子才能保持生长分化的状态;相对的,肿瘤细胞的增殖能力是自控、并且持久的,对于肿瘤细胞,在体外培养细胞的实验当中,无论是否添加外源生长因子,肿瘤细胞都能保持增殖分化的状态
- 肿瘤细胞可能能够自分泌生长因子,来模拟外源生长信号,进而达到恶性增殖的结果
细胞外生长信号的改变
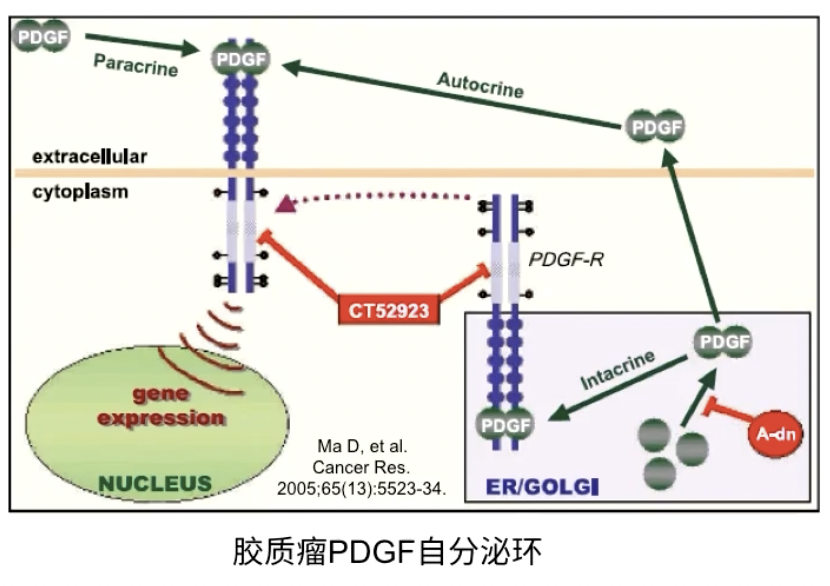
- 以胶质瘤为例,它是脑部最常见的肿瘤类型之一,预后很差,患者复发率高,这可能与胶质瘤细胞能够产生大量自分泌的细胞因子,促进其生长和增殖所致
- 其中一种胶质瘤细胞能够分泌的生长因子是PDGF,自分泌的PDGF与肿瘤细胞膜上的PDGFR结合,激活RTK信号通路
- 此外,在不同级别的PDGF当中,PDGF信号通路的配体和受体可能有不同程度的高表达,进而形成自分泌环路,持续刺激肿瘤细胞的生长和增殖
跨膜转导受体的突变
- 这些跨膜受体往往具有酪氨酸激酶活性,在与生长因子结合后,可以把生长因子传递的增殖信号传递到细胞内
- 在肿瘤细胞内,跨膜信号转导受体可能发生突变,尤其是高表达,进而促进细胞的生长,比如EGFR广泛分布于哺乳动物上皮细胞,成纤维细胞,胶质细胞,角质细胞等
- EGFR对于细胞的生长,增殖和分化等生理过程的发挥有重要的作用,EGFR在脑、胃、乳腺和部份肺部肿瘤中高表达
- 肿瘤细胞对生长因子变得敏感,引起更多的配体结合
- 激活配体非依赖的信号活化
细胞内信号通路的异常
- 最复杂的分子机制
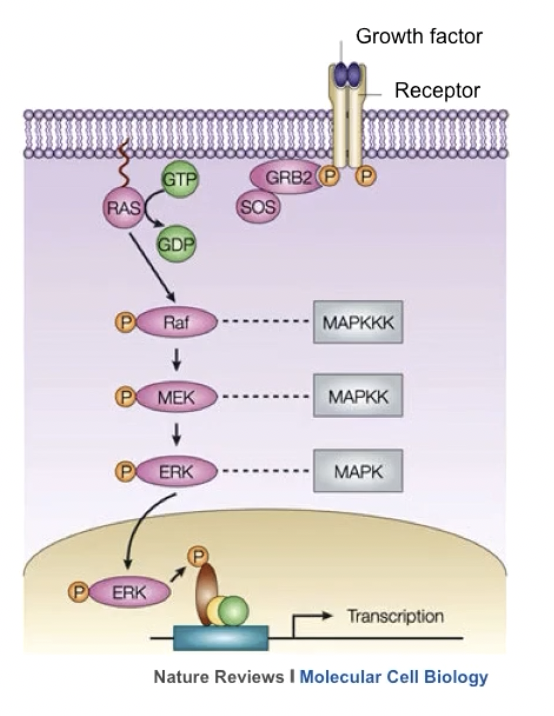
- 在大约25%的人类肿瘤中,原癌基因ras(也就是小G蛋白)发生突变,ras结合了GTP后会有磷酸化的活性,激活下游的信号通路
- ras是多种细胞信号转导途径的关键组分,其活性状态对细胞的生长和分化有重要的影响,在人类的肿瘤发生中,主要有k-ras、n-ras、h-ras这三种
- 癌基因ras突变后会导致ras蛋白在结构上发生改变,不需要上游信号的刺激就可以活化MAPK信号通路,导致肿瘤细胞的自分泌生长
哺乳动物的生长信号通路
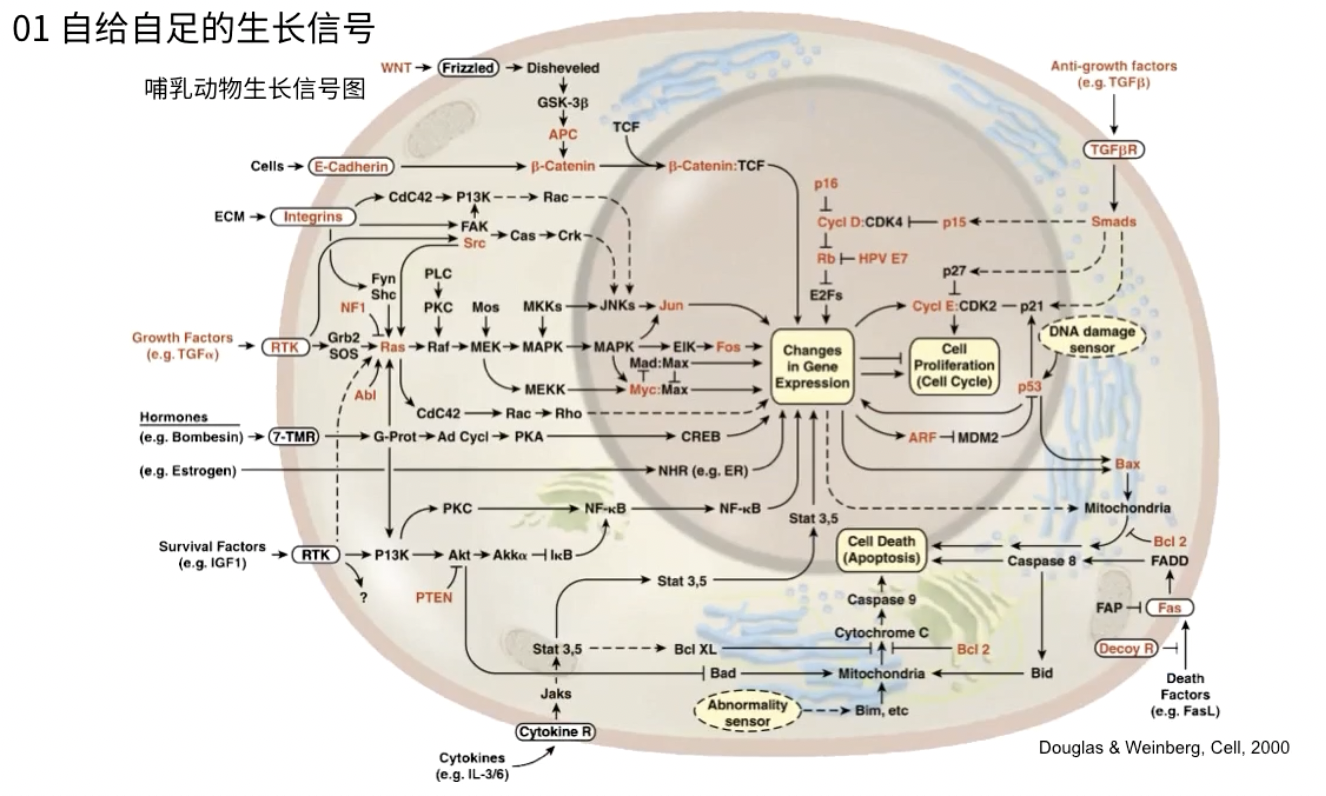
- 像是某种集成电路,以激酶为代表的蛋白就是这个集成电路上的晶体管
- 红色的分子是在肿瘤细胞中会异常表达的分子
- 常见的肿瘤信号通路
- Wnt
- NF-κB
- PI3K-Akt
- JAK/STAT
- TGF β
- MAPK
- 这些信号通路不是孤立存在的,它们相互之间通常形成交互网络,比如癌基因ras即可以激活MAPK信号通路,又可以作用于PI3K路径,同时影响细胞的生长
补充:异种细胞的相互作用
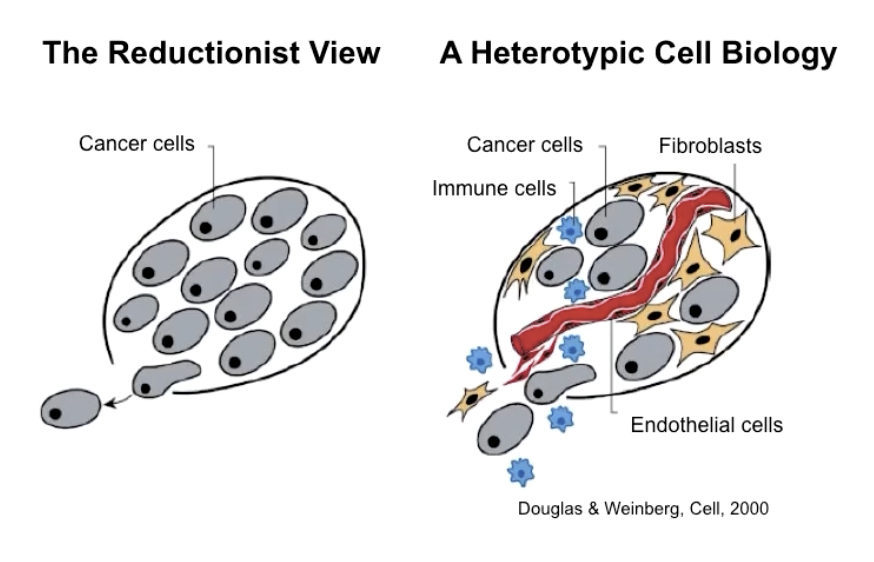
- 指的是肿瘤在发生发展的过程中,依靠肿瘤细胞与周围的正常组织细胞之间的异源相互作用。除了肿瘤细胞本身的改变之外,成纤维细胞、内皮细胞等基质细胞在促进肿瘤细胞生长的过程当中也起到了关键的作用
- 许多肿瘤细胞的生长信号来源是肿瘤的基质成分,基质细胞可以释放促肿瘤细胞生长的因子,在某些肿瘤中,炎性细胞可能可以促进肿瘤细胞的生长,而不是抑制其增殖
抗生长信号的不敏感/抗生长钝化
- 在正常的组织细胞内,除了存在生长信号外,还存在多种抑制信号,用来维持细胞稳态和自我平衡
- 这些抑制信号主要包括:可溶性生长抑制剂、细胞外基质抑制剂和相邻细胞表面的抑制剂
- 和生长信号传递的途径类似,抗生长因子也通过激活细胞膜受体,激活细胞内的信号通路
- 细胞抗生长信号途径的效应有两种
- 细胞从增长期进入静止期G0,在外界信号刺激下再返回增殖状态
- 细胞永远丧失增殖能力
补充:Rb基因/pRb蛋白的功能
- 主要讨论视网膜母细胞瘤蛋白,也就是Rb基因,蛋白写作pRb,是最早被发现的抑癌基因
- Rb基因定位于人类的13号染色体,编码的产物pRb定位于细胞核内,是细胞周期的负性调控因子
- Rb基因的作用主要依赖于pRb磷酸化的状态,高度磷酸化的pRb是没有活性的,低磷酸化的pRb有活性
- Rb蛋白还有两个同源的分子,p130和p107,但以Rb蛋白对细胞周期的调控最为重要
- Rb蛋白的作用机制
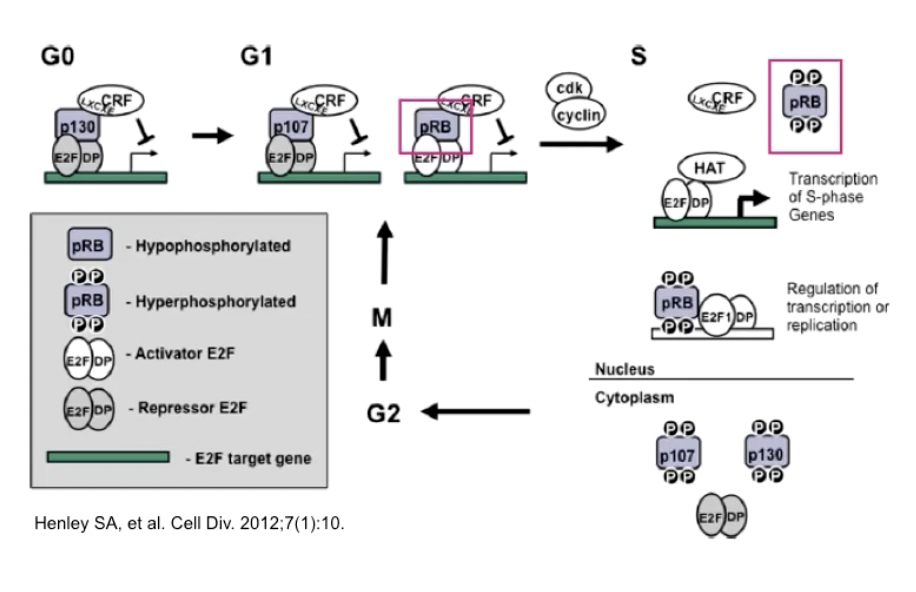
阻断抗生长通路
- 在肿瘤当中,Rb通路会发生一定的改变,使得转录因子E2F被释放,激活肿瘤增殖,导致细胞对抗生长信号不敏感
- Rb蛋白发生变化的内在机制
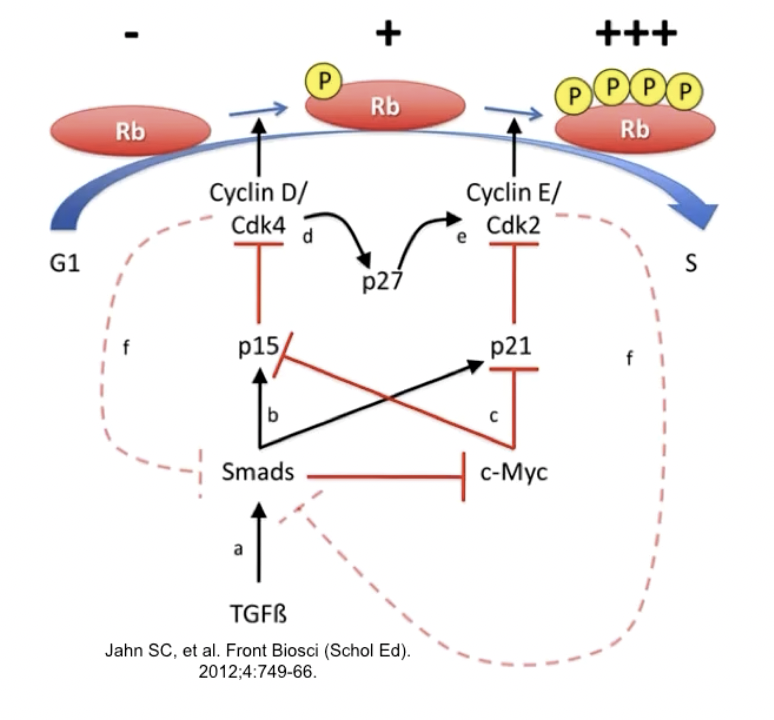
* TGF-β/SMAD通路可以调节pRb的磷酸化水平,进而调控细胞周期,然而在肿瘤中,这一通路的调控作用失控,造成肿瘤细胞对抗生长信号变得不敏感
* 图中TGF-β的效应:上调p15的活性,下调p21的活性,进而抑制细胞周期蛋白CDK4和pRb的磷酸化,达到抑制增殖的目的
* 肿瘤中可能发生的异常情况:TGF-β受体突变,SMAD,p15,CDK4和Rb基因的突变
* 总而言之,指向Rb蛋白的抗生长通路被阻断,导致肿瘤细胞对抗生长信号不敏感 - 其他的抗生长机制:在以宫颈癌为代表的DNA-virus诱发的病毒之中,pRb的功能可以被病毒蛋白清除,如下图所示
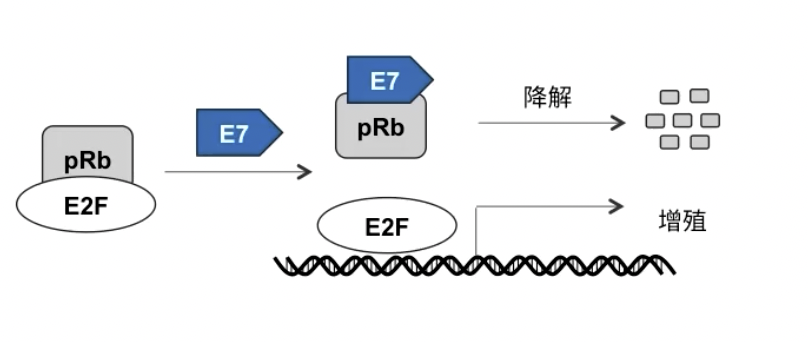
- 图中的E7蛋白来自人乳头瘤病毒HPV,产生竞争性抑制作用,而且与E7结合的pRb会逐渐被降解和清除
- 在阻断抗生长通路的过程中,肿瘤细胞可能会停止表达传递抗生长信号的分子,例如整合素和细胞黏附分子,导致肿瘤细胞对抗生长信号钝化
抑制细胞分化
- 典型的例子:Myc-Max-Mad网络
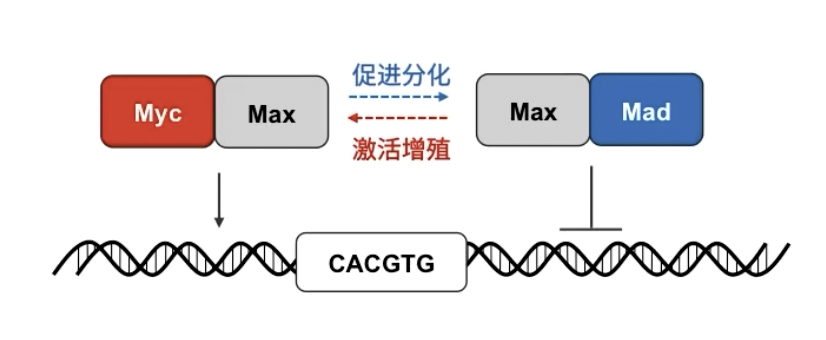
- Myc是原癌基因的一种蛋白产物,其通过与Max蛋白相结合,以二聚体的形式行使转录调节功能
- Myc-Max二聚体与DNA有很强的亲和力,可以特异性识别DNA序列中”CACGTG”的核心序列,并与之结合,激活分子的表达;最终表现出的生物学效应是促进生长
- 此外,Max还能与另一个蛋白Mad结合,这种异源二聚体同样识别DNA序列中”CACGTG”的核心序列,并与之结合,但起到的作用是转录抑制;最终表现出的生物学效应是促进分化
- 在肿瘤中,myc蛋白通常是过表达的,这会使得Myc-Max-Mad作用网络向着倾向于Myc的方向移动(也就是促进增殖、抑制分化)
逃避细胞凋亡
- 肿瘤细胞的恶性增长不仅仅由细胞增殖决定,也收到了细胞凋亡的影响
- 凋亡是细胞死亡的主要途径,而获得性抑制凋亡是肿瘤细胞的特点之一
细胞的凋亡途径
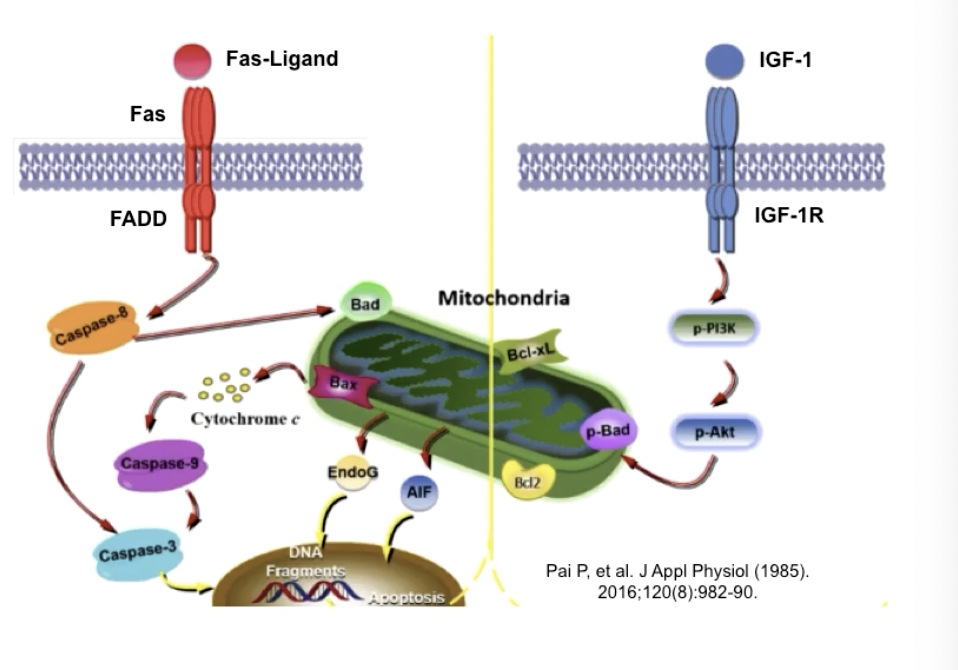
- 细胞膜受体类型的感受器
- 生存信号:IGF-1/IGF-1R,IL-3/IL-3R
- 死亡信号:FasL/Fas,TNFɑ/TNFR
- 细胞内的感受器
- 损伤
- 低氧
- 凋亡效应器
- Caspase-8
- Caspase-9
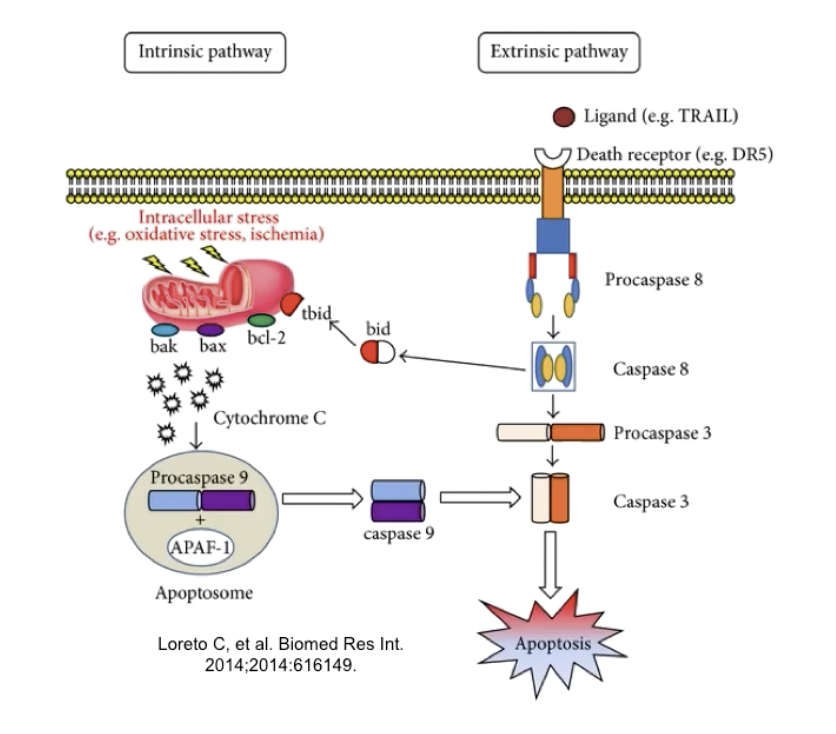
- 细胞凋亡的外源途径:受体-配体结合,通过Caspase的级联反应传递信号
- 细胞凋亡的内源途径:线粒体-细胞色素C途径
- 细胞应激-释放出CytC
- 凋亡小体出现(CytC-APAF-1-ProCaspase9复合物)-Caspase级联反应启动
肿瘤的抗凋亡机制
- 抑癌基因p53的突变
- wt-p53:上调Bax的表达,促进凋亡
- mutant-p53:功能丧失,细胞凋亡减少
- PI3K/Akt信号通路与肿瘤的抗凋亡
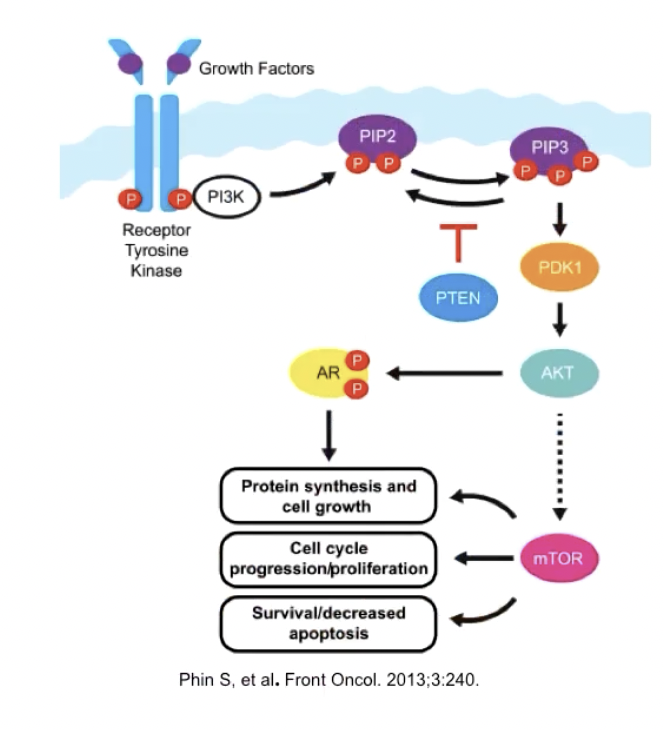
- PI3K由调节亚基p85和催化亚基p110组成,具有蛋白激酶的活性,可以被RTK激活
- PI3K通过两种方式被激活:与具有磷酸化酪氨酸残基的生长因子受体结合,引起二聚体的构象改变而激活;另一种是ras蛋白直接与p110调节亚基相结合,导致PI3K的活化
- 活化的PI3K催化第二信使PIP3的产生,进而改变Akt蛋白的活性,活化的Akt可以阻碍Caspase磷酸化,使其失活,进而抑制凋亡
- 在肿瘤细胞的胞外和胞内水平,可能会出现各种异常信号,异常激活PI3K/Akt信号通路,进一步抑制凋亡
- 胞外信号:IGF-1/IL-3表达升高
- 胞内信号:原癌基因ras的异常活化,PTEN基因的缺失
- 诱饵受体decoy receptor
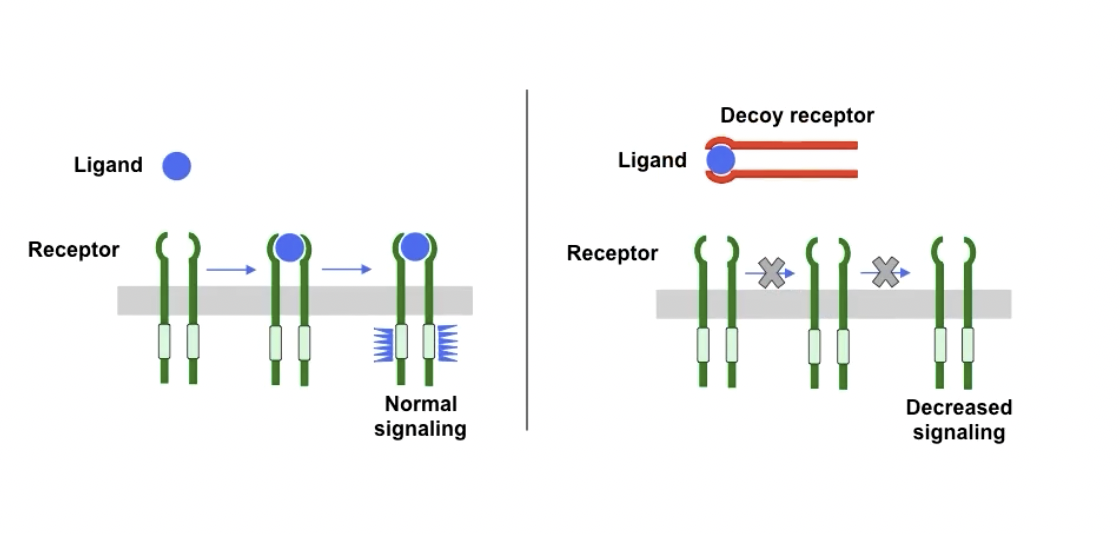
- “只结合配体,不执行功能“
- Fas的诱饵受体
- 下调死亡信号和抑制细胞凋亡
潜力无限的复制能力
Hayflick界限
- Hayflick界限:如果将细胞以1:2连续传代,子代细胞会逐渐出现衰老,退化和死亡,平均传代次数为40-60次,即使给予了丰富的营养,生长因子和生长空间也是如此,这个界限阐释了细胞的有限增长规律
- 原代细胞一般只能传50代左右,细胞的生长会出现停滞,大部份细胞会衰老和死亡,我们把这样的阶段称为”crisis”,也就是危机期
- 在肿瘤细胞中,以Rb和p53为代表的抑癌基因可能发生突变,诱导细胞突破“危机期”,获得无限增殖的能力
端粒&端粒酶
- 端粒:细胞有丝分裂的“时钟”和细胞寿命的度量衡
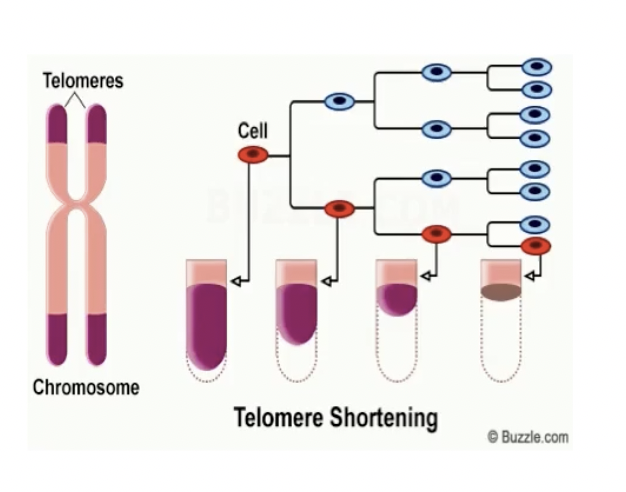
- 端粒是存在于真核细胞线状染色体末端的DNA-蛋白质复合体,由重复碱基”TTAGGG”组成,端粒就像是DNA的“帽子”,保护着遗传信息的稳定性
- 在细胞周期中,随着染色体的复制,端粒DNA会不断变短,细胞分裂一次,端粒DNA就会丢失50-100bp左右,一旦端粒DNA耗尽,细胞染色体就无法保持稳定,细胞将逐渐走向死亡,,因此,端粒的长度代表了细胞进行有丝分裂的潜力
- 端粒酶:细胞中特殊的酶,控制端粒的形成;端粒酶是RNA和蛋白质组成的复合体,其催化亚基利用自身的RNA作为模版,不断复制出端粒DNA,从而补偿染色体末端的复制消耗,保证染色体的完整性
- 端粒维持机制:几乎在所有的恶性肿瘤细胞中都可见,其中85-90%的肿瘤通过上调端粒酶的合成来修复端粒
- 复习二十四型 Lesson 2 细胞周期,Ink4A是一种细胞周期的负向调控蛋白,在小鼠中KO p16^Ink4A可以导致端粒酶表达增加,成瘤增加,反之亦然
- 其他的端粒维持机制:在其他10-15%的肿瘤细胞中,它们的无限复制能力不来自于端粒酶的活性,我们由此把这种机制称为端粒替代延长机制ALT
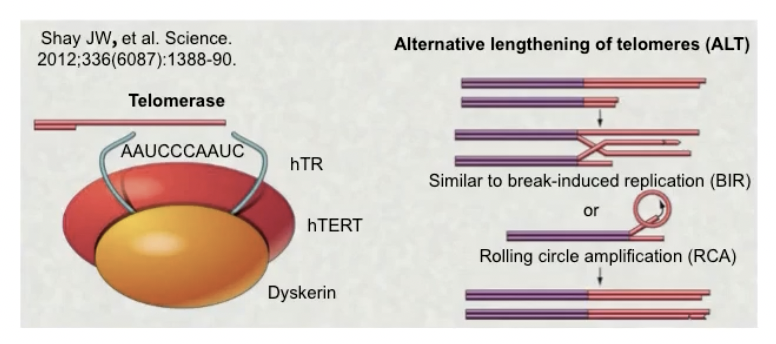
- ALT机制中,端粒的合成不依赖于端粒酶的RNA模版,而是以端粒DNA为模版,合成更长的端粒
持续的血管新生
- 处于异常增殖状态的细胞最初是缺乏生成血管的能力的,这样可以限制它们进一步向外扩张
- 为了达到较大的体积,肿瘤细胞不得不修炼“血管新生”的能力
促/抗血管生成因子
- 最常见的促血管生长因子是VEGF,血管内皮生长因子,具有促进血管通透性增加,细胞外基质变性,血管内皮细胞迁移,增殖和血管形成的作用
- VEGF家族主要有5种不同的生长因子,分别是VEGF-A-D和PIGF,VEGF通过不同的受体介导血管新生,目前发现的VEGFR包括VEGFR1-3,都是RTK,前两种主要在血管内皮细胞上表达,后者主要存在于淋巴管内皮细胞
- 受体与配体结合关系如下图所示:
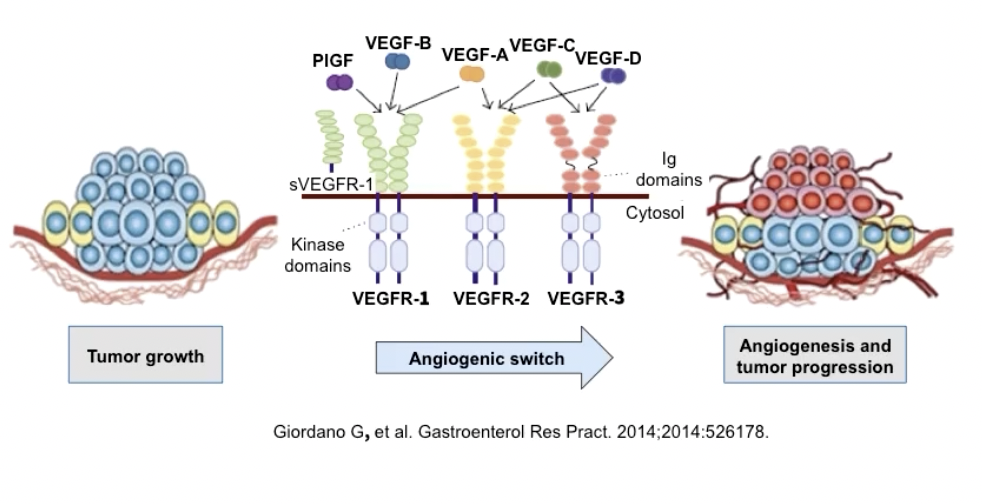
- VEGFR的主要功能是通过家族成员VEGFR-2来实现的,VEGFR-2与配体结合后形成同源二具体,使得胞内的酪氨酸残基磷酸化,进而执行血管新生的作用
- 在肿瘤治疗方面,抗VEGF抗体和抗VEGFR抗体已经被证明能够有效抑制血管新生和皮下肿瘤的生长
- 抗血管生成因子:TSP-1,从凝血酶刺激后的血小板细胞膜中分离的蛋白,广泛分布于各种组织(血液、心脏、肺部、软骨、大脑等)当中,在成纤维细胞,上皮细胞,内皮细胞,单核细胞中都有表达
- TSP-1能够与以CD36为代表的多种细胞表面受体结合,导致细胞内非受体依赖的酪氨酸蛋白激酶FIN磷酸化,进而执行抑制血管新生的功能
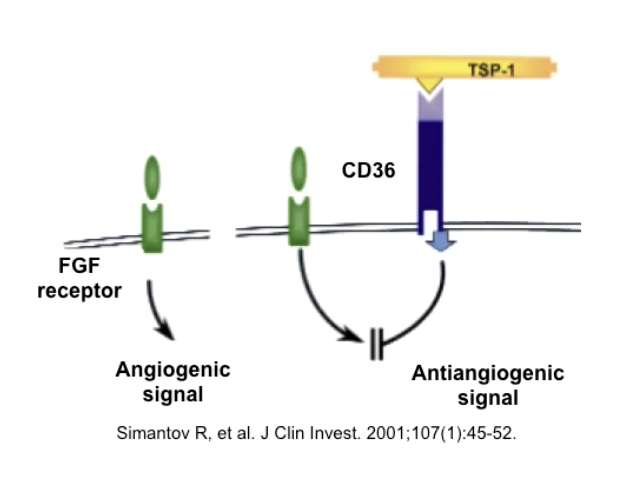
- 上图中的FGF是一种促血管生成因子,FGFR是一种RTK
肿瘤血管新生开关
- 肿瘤在发生发展的过程中,存在一种血管新生开关,揭示了肿瘤微血管形成的分子机制
- 在转基因小鼠中发现,小鼠的血管新生甚至早于肿瘤的形成,随后在人类的颈部、胸部和皮肤的癌前病变中也发现了血管新生的现象
- 肿瘤血管新生的过程中,涉及了促血管新生因子和抗血管新生因子之间的失衡,在这个过程中以VEGF和FGF为代表的促血管生成因子分泌增加,而以TSP-1和IFN-β为代表的抗血管生成因子相应地减少
- 在肿瘤生长最初阶段,并非所有的实体瘤都具备血管新生的能力;但是随着肿瘤细胞不断地生长和增殖、那些伴游癌基因和抑癌基因突变,并具备血管新生能力的肿瘤细胞逐渐在“进化”过程中,被自然选择形成增殖优势
血管新生调节机制
- 多种多样,抑癌明星基因p53可以上调抗血管新生因子TSP-1的表达,mutant p53失去功能,TSP-1表达量减少,释放肿瘤细胞的血管增殖潜力
- PTEN的作用与p53相似
- 促血管新生因子VEGF同样受到复杂的基因转录调控,比如原癌基因ras的激活可以导致VEGF表达升高
- 除了基因突变之外,蛋白激酶通过调节促血管生成因子或者抗血管生成因子的生物利用度,进一步控制血管新生的“开”和“关”
- 蛋白激酶可以释放细胞外基质中储藏的促血管生成因子FGF,另一方面,凝血系统的纤溶酶plasmin可以转变为血管抑素angiostatin,由原本的促血管生成转变为抗血管生成
- 肿瘤的血管生成机制提供了一种特殊的靶点,研制血管生成抑制剂,控制肿瘤的生成和转移将成为肿瘤防治的重要途径
组织侵袭和转移
- 在大部份肿瘤的发生和发展过程当中,或早或晚,原发肿瘤都会逐渐侵犯临近的组织,并通过血道或淋巴道到达主要的组织器官,形成新的转移灶
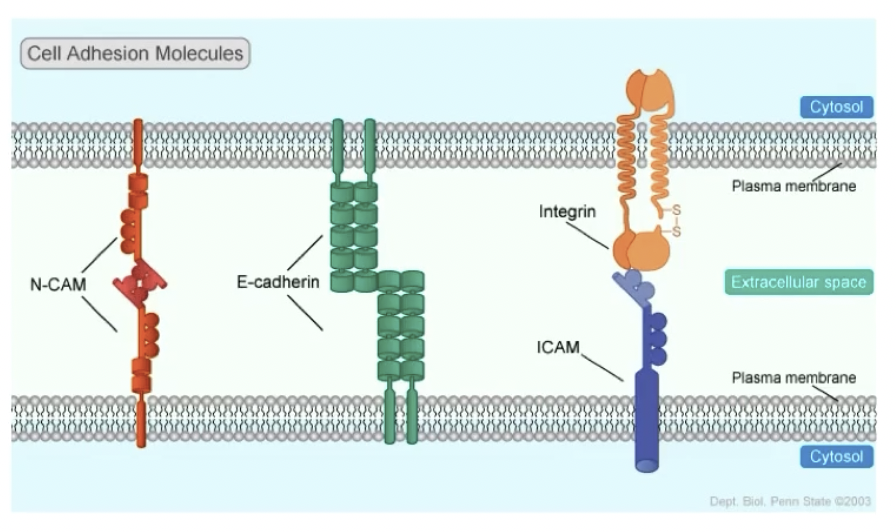
- Cadherin:Ca++依赖的细胞黏附素
- CAMs:细胞黏附分子
- Integrins:整合素
- Proteases:蛋白酶
- 黏附分子和黏附素是细胞与细胞之间,细胞与细胞外基质之间相互作用的膜表面糖蛋白,在肿瘤转移的每个环节中,都包含着黏附和解离两个主要的过程
- 整合素是介导肿瘤细胞受体与细胞外基质相结合的分子
- 蛋白酶的作用是破坏连接组织,降解细胞外基质,促进肿瘤细胞的迁移
Cadherin
- 跨膜糖蛋白家族,在钙离子存在时,可以抵抗蛋白酶的水解,主要介导同源细胞之间的连接
- Cadherin家族的主要成员:分布于上皮细胞的E-cadherin,分布于上皮和胎盘组织基底层的P-cadherin和分布于神经组织和肌肉组织的N-cadherin
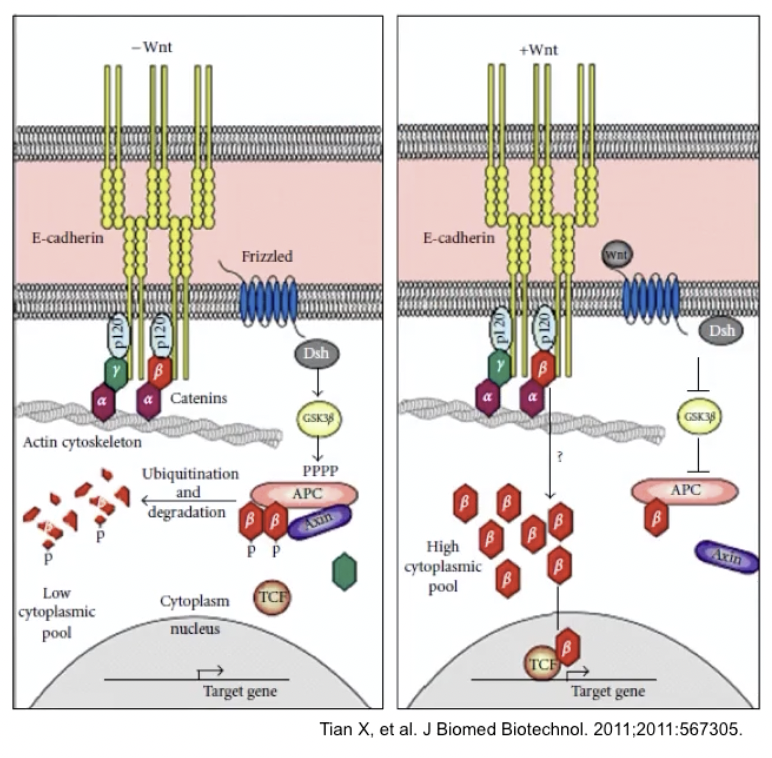
- 相邻细胞能够以E-cadherin作为桥梁,通过胞浆与β-catenin接触,经由Wnt通路传递生长或者抑制信号
- 在许多肿瘤中由于E-cadherin或β-catenin基因突变、转录失败或是细胞外水解等原因,导致内皮细胞E-cadherin功能缺失,对于体外培养的肿瘤细胞或转基因小鼠模型,过表达E-cadherin将降低转移和侵袭的倾向,反之亦然。
- E-cadherin能抑制肿瘤的转移和侵袭,E-cadherin功能丧失是肿瘤获得侵袭和转移能力的关键性步骤
CAM
- N-CAM是免疫球蛋白超家族分子,在肿瘤的侵袭和转移中起到了至关重要的作用
- N-neural cell,N-CAM介导了细胞与细胞间,以及细胞与细胞外之间的黏附来传导信号并且调控细胞的增长
- 有关N-CAM的研究显示,在神经细胞瘤、SCLC、侵袭性胰腺癌、CRC等肿瘤当中,N-CAM呈现低表达的状态,这使得肿瘤细胞由高度黏附形式转变为低黏附形式,甚至相互排斥,提示N-CAM异常的黏附形式对于肿瘤的转移具有重要的作用,转基因小鼠的实验支持这个结果
Integrins
- 整合素是一组细胞表面糖蛋白受体,由ɑ和β两个亚基组成
- 配体是细胞外基质ECM
- 整合素的主要功能是参与细胞与细胞之间的黏附,介导细胞与细胞外基质的结合
- 整合素在肿瘤发生发展的过程中具有双重性
- 一方面,在肿瘤发生的早期,整合素表达降低,可能导致瘤细胞与细胞外基质如基底膜等的黏附减弱,从而有利于肿瘤在局部的扩张
- 另一方面,当肿瘤细胞进入血液循环后,整合素表达升高,促使肿瘤细胞黏附于血管内皮,并进一步定位增殖
Protease
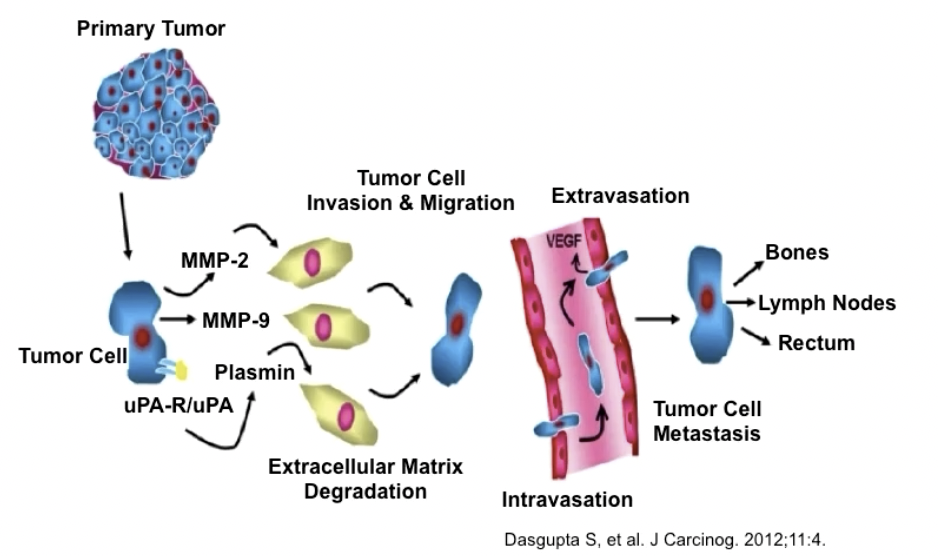
- 在很多肿瘤中,蛋白酶不仅可以从肿瘤细胞中产生,还可以从基质细胞和炎症细胞中获得,一旦被这些细胞释放,蛋白酶便被肿瘤细胞所运用
- 例如某种细胞在和基质细胞共同培养之后,能上调u-PA的表达,u-PA能和肿瘤细胞表面的受体相互结合,随后激活MMP系统,导致细胞外基质的降解
- 肿瘤的侵袭转移是临床治疗的最大障碍之一,侵袭转移本身是一个非常复杂的过程,主要包括黏附、降解、移动和增殖四个基本步骤,其中细胞与细胞或细胞外基质的黏附程度改变是肿瘤侵袭转移的始动步骤,而蛋白酶能够降解ECM的各种蛋白成份,破坏肿瘤细胞侵袭的组织学屏障,在肿瘤侵袭转移中起到关键作用
总结
- 总的来说,所有的肿瘤细胞都具有以上的6个获得性特征
- 但是,这些获得性特征在不同的肿瘤类型,以及不同发展阶段出现的顺序、以及频次是略有差异的
- 一般情况下,自分泌生长和抗生长钝化均出现在肿瘤发生的初期阶段,侵袭转移位居肿瘤侵袭的晚期,而逃避凋亡、无限复制和血管新生则出现在肿瘤发展的不同阶段
- 相应地,通过这些获得性特征得到的功能也有显著的差别,在某些肿瘤中,一种特异的获得性改变可能只是使其获得对应的单一功能,而在另外一些肿瘤中,可能诱发获取几种不同的功能
- 举例:突变的抑癌基因p53理论上同时具有血管新生和抵抗凋亡的功能,但是在不同肿瘤中,同样出现mutant p53,但是获得的功能不一定完全一样,甚至可以完全不一样
- 此外,多种原癌基因或抑癌基因的突变还可能导致某种肿瘤在不同的发生发展阶段,获得多次的同一种特征

