使用的文献:Long Noncoding RNA PICSAR Promotes Growth of Cutaneous Squamous Cell Carcinoma by Regulating ERK1/2 Activity

作者的前期成果
2003年
- 早在2003年,本文的通讯作者所在的实验室就发表了一篇Cancer Research,在这篇文章中,作者研究的模型是成纤维细胞Fibroblast,将成纤维细胞与p38的激动剂Arsenite孵育,成纤维细胞的存活率是明显下降的;进一步的机制研究发现,药物Arsenite显著激活了p38,但是在p38激活的同时,p-ERK受到抑制;已知p-ERK的表达升高可以促进细胞的增殖,所以研究人员提出p38会显著抑制ERK的磷酸化,进而抑制增殖,降低细胞的存活率
- 但是作者发现,这种在成纤维细胞内的,p38表达上调抑制ERK的磷酸化的机制,在肿瘤细胞中是不存在的;实验研究发现,在成纤维细胞HSF内,用TPA可以诱导MEK发生显著的磷酸化,在p38激活后,MEK的磷酸化水平是显著抑制的,说明在正常的成纤维细胞内,p38能够抑制MEK-ERK信号通路,但是在人的纤维肉瘤(HT-1080)细胞内,这种抑制是不存在的,也就是无论p38的激活状态,细胞内的p-MEK都处于高表达的状态,说明在恶性的HT1080细胞内,p38对于MEK/ERK信号通路是没有影响的
- 所以,作者在2003年的论文中提出的主要观点是:对于p38而言,其在正常细胞内可以抑制MEK/ERK信号通路,进而调节下游的细胞增殖;但是在恶变的细胞当中,p38对于MEK/ERK的抑制作用就消失了
2007年
- 这篇文章发表后的4年(2007年),作者又把他发现的这一机制从成纤维细胞和人的纤维肉瘤细胞,推广到了头颈鳞状细胞癌细胞。在07年的研究中,作者使用了42株HNSCC细胞系,这些细胞株分别来源于口腔、齿龈、喉和皮肤,在这些来源于皮肤的头颈鳞状细胞癌细胞系也被称为皮肤鳞状细胞癌细胞,其中的一部份细胞系也被运用到了2016年的研究中
- 所以,研究人员在2016年的研究中,大量采用了2007年的成果
- 在2007的研究中,研究人员在8株对照的人表皮角质形成细胞HEK和42株头颈鳞状细胞癌细胞中,检测了p38的4种异构体isoform(ɑ、β、γ、δ),在细胞内的表达检测水平发现,p38ɑ和p38δ在正常细胞系和肿瘤细胞中表达水平都较高,p38β和p38δ在正常细胞和肿瘤细胞内的表达都是很低的,同时,p38的磷酸化状态也会受到上游MKK的调控,MKK3和MKK6是调控p38的主要效应分子
- 在作者的研究中发现,在正常的人角质形成细胞和头颈鳞状细胞癌细胞中,几乎不表达MKK6,只表达MKK3
- 这些前期工作成果解释了为什么作者会选择在2016年的研究中重点关注p38ɑ和δ,以及MKK3这个分子了
- 通过机制研究,作者发现,在正常的角质形成细胞内,p38被激动剂Arsenite激活之后,p38的磷酸化水平显著升高,随着p-p38水平的升高,p-MEK/p-ERK的水平会受到抑制,这些结果提示,在正常角质形成细胞内,p-38负向调控MEK/ERK信号通路,这一结果和2003年的报道结果是完全一致的;但是在头颈鳞状细胞癌细胞内,检测到的结果是完全相反的,p38被激动剂Arsenite激活后,p-MEK/p-ERK的水平没有受到影响,这些结果说明,在恶性细胞内,p38失去了对MEK/ERK信号通路的调控作用
- 所以,综合2003和2007年作者研究的两项前期工作,作者发现在正常细胞内,p38对ERK有负向调控作用,正是由于这种负向调控作用的存在,使p38通过抑制ERK通路,达到抑制细胞增殖的目的,防止细胞发生癌变,但是在癌变细胞内,p38对ERK的抑制作用消失了,ERK不受到p38的调控,分子活性升高,使得肿瘤细胞发生不受控制的恶性增殖
本文工作详解
Fig.1 PICSAR的表达
- 在获得了筛选结果后,就涉及到了如何从筛选结果中挑选候选基因,进行深入研究的问题
- 通常的做法是筛选上调/下调2倍及以上的基因(Log2FC>1),但是很多时候获得的差异表达基因DEGs的倍数并不一定有2倍那么明显,所以有时研究人员也会筛选表达差异在1.5倍左右的基因(Log2FC=0.5849)
- 在这篇文章中,作者选择的是Log2FC>0.5,也就是表达差异>1.414倍以上的基因,由此也可以看出,在作者所研究的体系内,基因表达上调或下调的幅度没有那么明显
- 还有一些研究人员在选择差异表达基因时,会考虑p-value,其产生是因为采用了t-test的方法,检验差异表达基因的表达,通过合并样本间可变的数据,来评价差异表达,用于评价某一基因在两组样本中的表达是否具有差异;由于芯片或者测序的成本非常高,所以样本量通常比较少,对总体方差的估计是不太准确的,t-检验的检验效能会降低,检验效能是不全面的,为了提高检验的效能,研究人员又开发出了SAM算法,纠正多重假设检验中的假阳性率,q-value恒量的是某一个基因假阳性的概率,显然如果q-value越低,那么出现假阳性的概率也就越低,可验证性越高
- 特别强调,无论是p-value还是q-value,或是其他的检验方法,本质上都是研究人员开发出来的数学算法,但是生物学是一门实验的科学,只有通过实验可以验证的结果才是可靠的。需要避免盲目、过度地依赖生物信息学,实验验证才是王道
- 另一种可以提高筛选效率的方法是采用两轮筛选法,即上一讲组织芯片的时候提到的方法,这种方法对于功能基因和编码基因来说比较有效,因为组织芯片从原本上来说取材于石蜡组织块,因此可以用组织芯片进行功能基因的免疫检测,进行二次筛选;但是普通的石蜡组织在制备和取材过程中没有经过特殊的处理,因此容易导致组织内的RNA降解,所以不适合也不推荐用组织芯片进行LncRNA候选分子的二次筛选
- 研究人员在通过RNA-seq获得显著差异基因PICSAR之后,在细胞系和临床组织样本中进行了PICSAR的表达验证,验证的结果证实了PICSAR在皮肤鳞状癌细胞系和皮肤鳞状细胞癌临床样本中都有高表达,验证了筛选的结果,但让人感到稍有遗憾的是作者仅在6例临床样本中进行了PICSAR的检测,临床样本的数量偏少
- 需要注意的是,在Fig.1B中,作者分别在正常细胞系,原位皮肤鳞状细胞癌细胞系和转移性皮肤鳞状细胞癌细胞系中检测了PICSAR的表达水平,仔细分析数据可以发现,PICSAR在原位皮肤鳞状细胞癌细胞中的表达较高,但是在转移性皮肤鳞状细胞癌细胞系中的表达又下降了,呈现出正常细胞低表达,原位癌高表达,转移性癌又降低的特点,这一结果和RNA-seq中筛选的结果并不完全一致,但是作者通过将原位皮肤鳞状细胞癌细胞系和转移性皮肤鳞状细胞癌细胞系的数据放在一起展示,比较巧妙地规避开了这一点,这种展示数据的小技巧值得借鉴
- 另一个需要注意的,Fig.1B中qPCR法检测PICSAR,发现在原位癌中高表达,但是转移性癌细胞系内的表达降低,这一结果和Fig.2的结果是有所出入的,因为Fig.2的结果显示的是PICSAR的表达会随着肿瘤恶性程度的增高而提升,可能的解释1)Fig.1b里用的是细胞系,而Fig.2用的是临床样本,两种样本来源的差异可以解释。2)尽管表达PICSAR的数目随着肿瘤的进展而升高,但是每个细胞内表达的PICSAR分子是有可能降低的,造成的总效应是PICSAR的总表达水平随肿瘤进展而降低
- 最后,在Fig.1d中,作者通过ISH的方法,研究了PICSAR在皮肤鳞状细胞癌细胞系内的亚细胞定位和分子在移植瘤组织当中的表达,在皮肤鳞状细胞癌细胞内,免疫组化显示,PICSAR主要分布在胞浆中,LncRNA的亚细胞定位对于其功能的预测具有一定的意义,如果LncRNA分子主要分布在细胞浆内,则提示这个分子一般不会通过调节基因转录的方式调节下游的信号通路;作者在全文的讨论部份也推测,PICSAR有可能在胞浆内,通过直接或间接的作用,调控DUSP6的mRNA,但具体的作用机制,本篇文章没有详细的研究
- Fig.1e做的是移植瘤模型,在移植瘤模型中,作者使用的探针同时可以通过碱性磷酸酶显示出可见的红色,又可以发射出荧光,所以在Fig.1e中左侧的红色点和右图的红色荧光是有共定位的
Fig.2 PICSAR在cSCC中高表达,而且与临床分期密切相关
- cSCC:cutaneous squamous cell carcinoma,上皮鳞状细胞癌
- 作者在Fig.2中,选取了不同病理阶段的组织样本,并检测了PICSAR分子的表达;作者选取了4种临床病理类型,分别是正常皮肤,紫外线诱导的癌前病变,原位皮肤鳞状细胞癌和浸润性皮肤鳞状细胞癌
- ISH显示,在正常组织中几乎没有PICSAR的表达,但是随着疾病的不断进展,表达PICSAR的细胞比例不断上升
- Fig.1和Fig.2都是非常经典的套路,首先通过高通量筛选,获得差异表达基因,随后在细胞系和临床组织样本中进行差异表达分子的验证,尤其是Fig.2证实PICSAR阳性细胞的比例随着疾病的进展不断升高,证实PICSAR的表达水平具有临床意义
- 唯一让人觉得遗憾的是,检测的临床样本数目不是很多,所以PICSAR作为皮肤癌诊断的临床标志物的意义还需要更多的研究支持
Fig.3 p38负调控PICSAR的表达
- 从这里开始,作者开始深入研究PICSAR及其所介导的信号通路
- 从一开始,研究人员就把信号通路锁定到了p38和ERK/MAPK信号通路,可能的原因有以下两点
- 在皮肤鳞状细胞癌中,可以检测到高频率的h-ras/k-ras以及EGFR信号突变,这些基因包括ras和EGFR等,都是ERK/MAPK信号通路中的关键分子
- 作者从2003年开始就一直在研究p38和ERK/MAPK信号通路
- 作者首先采用抑制剂阻断的方式,研究在细胞内ERK或p38被阻断的情况下,PICSAR的表达水平是否受到影响,作者选的抑制剂有3种(见下),发现在PD药物阻断ERK通路和SB药物阻断p38ɑ和β时,PICSAR的表达是不受到影响的,只有在BIRB阻断了p38的全部4种异构体后,PICSAR的表达才显著升高

- 随后,在细胞内,通过RNAi的方法,敲减了p38ɑ和p38δ,证实单独敲减p38δ或同时敲减p38ɑ和p38δ都可以提升PICSAR的表达
- 最后,在Fig.3c中,研究人员在细胞内高表达了MKK3的无功能形式(dominant negative 质粒),阻断了MKK3,进而阻断了p38的信号转导,同样可以显著提升PICSAR的表达水平
- 但是,在这部份的实验中,有以下几个问题是值得商榷的
- 作者为什么选定了CREB作为p38下游的效应分子?根据现有的研究证明,p38可以影响包括CREB在内的许多转录因子,并引起下游整个转录调控网络的变化,可以认为CREB是p38下游的效应分子;但是ERK也可以影响包括CREB在内的许多转录因子,同样可以引起下游整个转录调控网络的变化,所以在进行实验之前,作者是根据什么认为CREB是p38下游的效应分子,而不是ERK下游的效应分子的?
- 当然,通过实验结果我们可以反向推断,在PD药物阻断了ERK信号通路后,CREB的表达没有降低,但是在用BIRB和SB药物处理后,阻断了p38信号通路,CREB的表达就显著下降了,因此,反向推导出CREB是p38的下游分子,因为CREB的表达收到p38抑制剂的影响
- 更严谨的验证方式应该是和PD药物阻断ERK信号通路,检测ERK的磷酸化水平类似,通过p38的异构体特异性抗体,进行免疫共沉淀IP实验,富集p38的各种异构体,然后在富集的p38异构体水平中,检测异构体的磷酸化水平,证实不同抑制剂对于p38的异构效率,但这样做工作量就显著增大了
- 第二个需要讨论的问题:根据现有的研究报道,ERK下游调控CREB,进而调控细胞恶性增殖,已经形成了一条经典通路,但是在作者研究的实验体系内,ERK下游不依赖CREB,由此引出的问题是在作者研究的皮肤鳞状细胞癌模型中,ERK到底通过哪一种CERB非依赖的方式,介导了肿瘤的恶性增殖?但可惜,本文作者没有深入研究
- 第三个需要讨论的问题:在Fig.3a中,作者发现用BIRB或者SB药物阻断了细胞内的p38信号通路后,对于ERK的磷酸化水平没有影响,这一结果和作者2003以及2007年的前期工作是一致的,因为作者发现p38只有在正常细胞当中,才会抑制ERK信号通路,在癌变的肿瘤细胞中,p38是没有对ERK的调控作用的;因而我们可以得到结论,p38阻断之后,PICSAR的表达水平是升高的,但是不影响ERK的表达水平,换言之,PICSAR的表达水平不影响ERK,但是在文章后续的研究过程中,作者得出了敲减PICSAR之后,ERK的磷酸化水平是降低的,由此得到的结论是PICSAR可以正向调控ERK的磷酸化水平,这两个结论是明显冲突的,而作者没有给出合理的解释
- 第四个问题:用SB药物处理了p38ɑ和β后,PICSAR的表达没有发生显著变化,从这个结果中已经可以排除p38ɑ和β对PICSAR的作用,而且由于研究人员在2007年的文章中已经证实了,p38β和p38γ在鳞状细胞癌中几乎不表达,因此我们可以得到的结论是,PICSAR的表达仅仅受到了p38δ的影响,但是作者在RNAi实验中,一定要检测p38ɑ和p38δ,我们推测这样可能的原因是1)作者想要保证2016年的研究结果和2007年的研究结果在形式上有一致性2)单独敲减p38ɑ对实验结果虽然没有显著影响,但是当p38ɑ和p38δ同时敲减之后,具有一个显著的协同效应
Fig.4 沉默PICSAR抑制cSCC细胞增殖和转移
- 作者在Fig.4的研究中,研究了PICSAR对于表型的影响,作者首先设计了针对于PICSAR的特异性siRNA序列,并且在三株不同的细胞中,验证了敲减的效率;ISH的结果也显示,在对照条件下,是可以在胞浆内检测到PICSAR的阳性信号的,但是当PICSAR被特异性敲减之后,细胞胞浆内的PICSAR信号就几乎完全消失了,说明RNAi的敲减效率是非常显著的
- 一个值得注意的问题:作者应用了3株细胞进行敲减验证,但是对照组只有1个,理论上说实验组有3组,对照组也应该有3组
- 在Fig.4b中,研究人员发现,在PICSAR敲减之后,细胞的存活率明显下降,说明PICSAR在细胞中可以起到类似癌基因的作用,wb证实,PICSAR下游是可以通过ERK信号通路介导肿瘤细胞的增殖的,因为PICSAR敲减之后,细胞内p-ERK的水平明显降低了;作者在这里获得的结论是PICSAR可以正向调控ERK
- 通过划痕愈伤实验,作者发现PICSAR敲减后可以显著抑制皮肤鳞状细胞癌细胞的迁移,同样说明PICSAR在皮肤鳞状癌细胞中可以起到类似癌基因的作用,促进转移
- 在生物学研究中,用于验证肿瘤细胞转移的实验有很多,除了划痕愈伤实验以外,其他的还有transwell转移小室实验,通常情况下为了证明肿瘤细胞的迁移实验,可以同时使用上述的两个实验,或者单独使用transwell实验,不推荐单独使用划痕愈伤实验,因为在划痕愈伤中,孵育过程通常比较长,因此肿瘤细胞存在增殖的效应,仅仅通过划痕愈伤实验没有办法排除肿瘤细胞增殖带来的影响
- 另一个需要注意的是,尽管实验已经证实,敲减PICSAR之后,细胞内的p-ERK水平降低,但是,研究人员没有做rescue实验,这是一个小小的瑕疵
- 一个两难的境地
- 一方面,如果根据本文的内容,认为p38负向调控PICSAR(Fig.3),而PICSAR又可以正向调控ERK得磷酸化,那就等于承认p38可以间接调控ERK
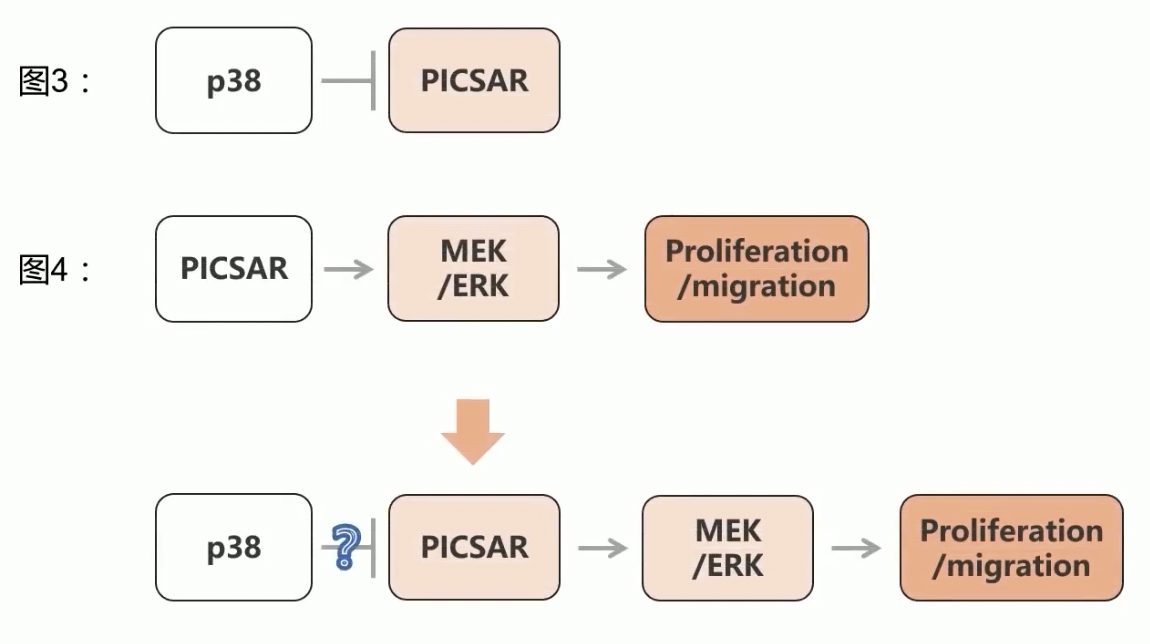
- 但是这样一来,就和Fig.3a的结果是矛盾的,和作者2003年以及2007年发表的结果也是矛盾的,因为这俩发现,在癌变的细胞中,p38对ERK没有调节作用,如果认可了Fig.3a的结果,也就是抑制了p38可以提升PICSAR的表达水平,但是对于ERK的表达水平没有影响,那又如何解释,Fig.4中PICSAR敲减之后抑制了ERK的表达水平呢
- 作者在这里获得的结果和之前的内容,包括2003和2007年以来的结果,是有明显的矛盾的;从后续的研究结果来看,研究人员还是认可PICSAR能够调控ERK的表达,并且对如何调控的机制做了研究
Fig.5 PICSAR调控DUSP6/ERK信号轴
- 接下来,为了研究PICSAR下游对于ERK的调控通路,研究人员再次通过转录组测序,比较了对照条件和PICSAR敲减条件下,发生显著变化的分子,筛选条件为Log2FC>0.5,同时p值<0.05,筛选出这些基因后,再进行Ingenuity Pathway Analysis,这个分析是通过对应的IPA软件进行的
- 研究结果发现,在PICSAR敲除后,和细胞周期、肿瘤恶变以及翻译后修饰的通路都发生了显著的变化,GO分析和KEGG通路分析提示,在PICSAR敲减后,和细胞增殖以及细胞转移相关的通路都发生了显著变化,这些通路包括cell proliferation,细胞迁移regulation of cell migration,细胞外基质结合Extracellular binding,细胞外基质受体相互作用ECM reeptor interaction等等
- 通过RNA-seq,研究者分析了表达调控最为显著的分子,这些分子中,研究者发现PICSAR敲减后,DUSP1是上调最为明显的分子之一,DUSP1又叫MKP1,可以去磷酸化p38/JNK,但是在作者的研究中, PICSAR敲减后不影响p38的表达,因此实验结果不支持PICSAR敲减导致DUSP1升高,进而调控p38的这条信号通路,所以作者无奈之下只能研究DUSP6,也就是MKP3(背景中有介绍DUSP6可以去磷酸化ERK,而ERK又受到了PICSAR的调控,所以是可能存在PICSAR通过调控DUSP6来实现对ERK的调控的)
- 通过RNA-seq分析,DUSP6在PICSAR敲减后表达上调了2.13倍,上调倍数明显,在皮肤鳞状细胞癌细胞系12A和59A中,在PICSAR敲减以后,可以检测到DUSP6的mRNA和蛋白水平都显著升高,提示PICSAR敲减后上调了DUSP6的表达,而且PICSAR敲减后p-ERK水平的降低依赖于DUSP6的介导,因为使用BCI药物阻断了DUSP6之后,无论PICSAR是否敲减,ERK的磷酸化水平都不会受影响,说明PICSAR通过DUSP6调控ERK的磷酸化,Fig.6f是经典的rescue实验
- 由此,研究人员获得结论,PICSAR通过DUSP6来调控ERK的磷酸化水平
Supplementary S5 PICSAR调控下的ERK信号通路
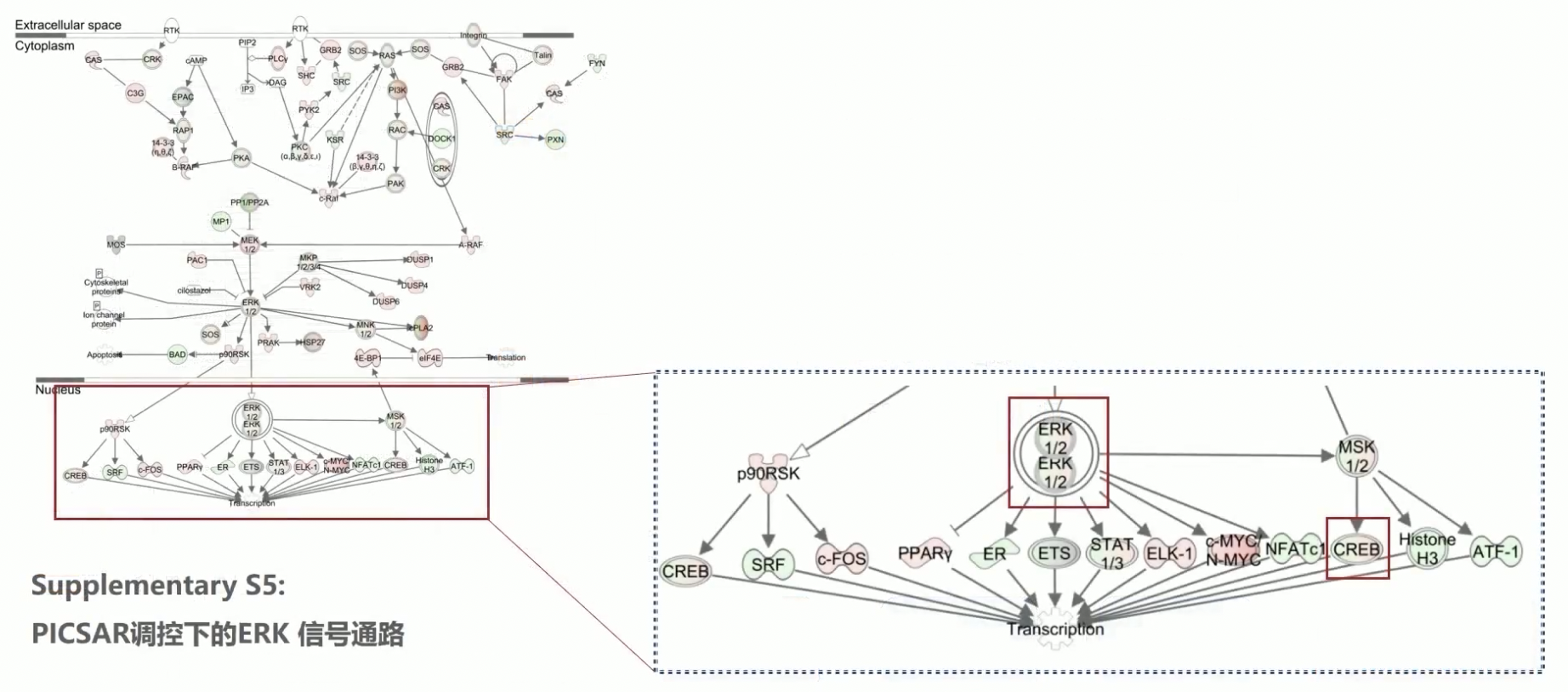
- 通过IPA软件,研究人员还研究了PICSAR敲减后的,ERK信号通路的网络构架图,这部份数据作者展示在了Supplemental Data中
- 这张网络图中,红色表示上调,绿色表示下调
- 通过分析可见,PICSAR敲减后,ERK表达下调,这一结果和Fig.4c的结果是一致的,但是ERK下游的CREB表达也下调,也就是PICSAR敲减后下游的ERK和CREB的表达都会降低,这个结果和Fig.3a是矛盾的,因为那张图中展示的是在用PD药物阻断了ERK之后,CREB的表达不仅没有下调,反而变高了
- 另一个矛盾点:根据Fig.3a,PD药物阻断了ERK信号通路,不影响CREB,所以研究人员把CREB作为了下游的效应分子,如果根据RNA-seq的筛选结果构建的IPA通路图进行参考的话,ERK下游影响了CREB,那么把CREB作为p38的下游效应分子就是不恰当的了,因为CREB的表达同时受到了p38和ERK的共同调控
Fig.6 沉默PICSAR抑制cSCC的成瘤能力
- 研究的是PICSAR的在体功能
- 通过移植瘤模型,PICSAR敲减的cSCC细胞所形成的移植瘤体积会显著小于对照组形成的,在实验完成后,取出移植瘤称重,结果也显示,对照形成的移植瘤重量显著大于PICSAR敲减的组别
- 通过移植瘤组织形成的免疫组化结果,证实PICSAR敲减的组别中,Ki-67的阳性细胞比例显著降低,这个结果也与PICSAR敲减后移植瘤体积减小,增殖减缓的现象
- 如果作者在移植瘤中再检测DUSP6的表达,以及ERK的磷酸化水平,那么实验数据会更加详实
总结
- 作者的主要发现是在皮肤鳞状细胞癌细胞中,发现一个特异性表达升高的LncRNA分子,并且把这个分子命名为了PICSAR
- 机制研究发现,PICSAR上游受到p38δ的调控,下游又可以通过DUSP6调节ERK的磷酸化水平,同时PICSAR可以在皮肤鳞状细胞癌内起到类似癌基因的作用
- 但是,整篇文章数据解构下来,我们还是会发现,有一些问题值得我们的思考

- 作者在做机制研究的时候发现,PICSAR上游受到p38δ的调控,下游通过DUSP6影响ERK的磷酸化水平,带来的问题是如果同时采信这两项结果,就可以得出p38δ可以间接调控ERK磷酸化水平的推论,而这个推论是和作者在2003以及2007年的结论相违背的;而如果不认可这个言论,又没有办法理解为什么p38与ERK的相互调控关系不成立,这篇文章的结论和作者之前的研究成果存在比较大的矛盾
- 作者研究了本质是LncRNA的PICSAR在细胞内的表型,可以起到类似癌基因的作用,因为PICSAR敲减后可以抑制肿瘤的增殖和转移;同时,机制研究发现PICSAR下游可以影响ERK的磷酸化水平,但是不够严谨的地方在于,作者没有在他的研究体系内再一次通过rescue实验证实,PICSAR敲减之后,可以通过过表达p-ERK来rescue PICSAR的表型,在这方面,作者的研究是有缺憾的
- 过度追求研究结果和之前研究在形式上的一致性:作者在2007年的研究中,发现p38ɑ和p38δ在头颈鳞状细胞癌中是有作用的,而且表达也比较高,所以作者在本项研究中也略带有偏执地把p38ɑ和p38δ扯在一起,实际上通过SD药物的阻断功能实验已经可以证明p38ɑ对PICSAR的表达是没有影响的
- ERK介导的CREB非依赖的增殖通路没有足够的深入:虽然很多文章都报道,ERK可以促进肿瘤细胞的增殖,但是绝大部份的报道都着重于 CREB依赖的ERK作用途径,所以ERK-CREB-肿瘤信号增殖已经成为经典通路了,但是在这项研究中,作者发现PD药物阻断了ERK后,ERK下游的CREB表达没有下降,也就是说ERK通过了一条CREB非依赖的途径来调控细胞增殖的,但是这方面的研究在本项研究中没有深入,难免让人觉得有些遗憾。


