使用的文献:Thyroid transcription factor 1 enhances cellular statin sensitivity via perturbing cholesterol metabolism

- 在这一节内容中,我们主要关注以转录因子为靶点的药物开发现状,这也是转录因子研究目前最为热门的潜在应用领域之一。
- 转录因子广泛存在于机体各种组织细胞中,目前在人类基因组中已经发现了超过2000个转录因子。转录因子对多种信号分子,比如细胞因子、生长因子、细胞黏附因子、抗凋亡蛋白基因的调控等都起到关键的作用。而且,转录因子还在多种疾病,包括肿瘤等恶性疾病的发病机制中发挥重要的作用。转录因子的研究具有重要的临床意义和潜在的应用价值,不仅仅是因为很多转录因子的突变或者失调是导致疾病发生的重要原因,更重要的是,转录因子可以作为药物靶点,一些小分子化合物能影响转录因子的二聚体形成,或者影响转录因子与DNA分子的结合。因此,它们可以改变转录因子介导的基因调控。
已获得FDA批准的药物
- 已经有针对转录因子的药物获得了FDA的批准并用于相关疾病的治疗,有两个很好的案例,一个是他莫昔芬,另一个是比卡鲁胺。
他莫昔芬
- 他莫昔芬,又名三苯氧胺,是一种甾体类抗雌激素药物,目前被广泛地用于乳腺癌的治疗,是绝经前乳腺癌术后内分泌治疗的标准药物。它能够降低雌激素依赖型乳腺癌患者的死亡率和复发率,具有良好的临床效果。
- 他莫昔芬在肝脏内代谢,在细胞色素P450,也就是CYP酶家族的作用之下,他莫昔芬代谢为N-去甲他莫昔芬。N-去甲他莫昔芬在细胞色素氧化酶2D6,CYP2D6的作用下,继续代谢为4-羟基-N-去甲基他莫昔芬,以及4-羟基-他莫昔芬。其中,4-羟基-N-去甲基他莫昔芬是主要的代谢产物,在对抗雌激素治疗中起着极为重要的作用。
- 4-羟基-N-去甲基他莫昔芬可以和细胞内的雌二醇竞争性地结合雌激素受体,也就是ER受体,形成他莫昔芬-雌激素受体的复合物,改变雌激素受体的空间构象,使得雌激素受体的DNA结合区域不能暴露,也就阻断了雌激素受体和靶基因转录调控区域的结合,从而达到阻断雌激素对下游基因转录调控的作用。
比卡鲁胺
- 另一个被FDA批准用于临床治疗的药物是比卡鲁胺。比卡鲁胺也是一种非甾体类雄激素结抗剂,可以在肿瘤部位和雄激素竞争性结合雄激素受体Androgen Receptor AR,目前已广泛地应用于前列腺癌的临床治疗。
有前景的转录因子抑制剂
- 除了他莫昔芬和比卡鲁胺已经获得FDA批准,广泛用于临床治疗之外,还有多个针对转录因子及其信号通路的化合物,比如NF-κB通路抑制剂和STAT通路抑制剂,已经获得良好的临床前研究结果,并且开始了相关的临床验证。
NF-κB抑制剂
- NF-κB蛋白家族成员广泛存在于各种真核细胞内,也是哺乳动物中研究比较多的一类转录因子。很多研究结果证实NF-κB是应激和炎症反应的关键调节因子,参与细胞增殖和凋亡等过程,在肿瘤发生和发展过程的信号传递中发挥重要作用,与肿瘤细胞的侵润、转移也密切相关。
- NF-κB不是一个分子,而是一个家族,包含5个成员。我们通常说的NF-κB,指的是形成二聚体化的蛋白复合物,通常是指p50/p65二聚体。
- NF-κB信号通路,我们已经在上一讲内容中介绍过,这里简要回顾一下:
- 当通路没有激活时,NF-κB在胞浆内和i-κB相互结合。i-κB通过与NF-κB结合的方式,起到NF-κB抑制剂的作用,抑制了NF-κB的功能。
- 当NF-κB信号通路被激活之后,IKK激活,磷酸化i-κB,并且导致i-κB最终被细胞内的蛋白酶体降解,使得NF-κB重获自由,并转位到细胞核内,激活了含有NF-κB的DNA结合位点的基因表达。
- 近年来,针对NF-κB抑制剂的设计和研究已经取得了大量的研究成果和实践工作。目前处于临床和临床前实验的抑制剂主要可以分为四大类。
天然产物
- 代表性的有小白菊内酯、姜黄素、染料木素等。
- 小白菊内酯是一种倍半萜烯内酯内酯化合物,是植物药野柑菊的主要活性成分。近年来,多项研究发现,小白菊内酯可以诱导白血病、多发性骨髓瘤等多种肿瘤细胞的凋亡,同时对其他抗癌药物有增敏效果。
- 小白菊内酯是一种有效的核转录因子NF-κB激酶的抑制剂阻断细胞内的基因表达调控,促进氧化应激,产生基因毒性作用,还能通过使p53前体磷酸化,激活p53介导的细胞周期阻滞现象。
- 研究表明,小白菊内酯可以抑制白血病干细胞增殖,并且使细胞停滞于G2期。但由于小白菊内酯成药性比较差,限制了它的临床应用,所以小白菊内酯的类似物二甲氨基内酯应运而生。
- 二甲氨基内酯缩写就是DMAPT。DMAPT具有良好的药理活性,同时在自发性急性白血病、小鼠的模型体内实验中具有良好的效果。目前,DMAPT已经在英国开展了临床试验。
合成类小分子
- Bortezomin,中文名硼替佐米、MG-132等。
- 硼替佐米是美国FDA批准的第一个蛋白酶体抑制剂,用于治疗初发或复发性难治性多发性骨髓瘤的一线药物。硼替佐米抑制蛋白酶体的机制是通过阻断26S蛋白酶体活性,抑制i-κB和NF-κB的解离,使NF-κB无法入核调控下游基因的转录,影响细胞内多种信号级联,导致肿瘤细胞生长延迟或者死亡。
- MG-132同样是一个有效的蛋白酶体抑制剂。其机制和上面的硼替佐米类似,也是通过抑制调节分子i-κB的降解,维持i-κB表达的高水平,从而抑制NF-κB的活性,使得NF-κB驻留在细胞浆内,始终处于无活性的状态。
细胞膜穿透性肽类
基因治疗方法
STAT通路抑制剂
- STAT全称是Signal Transducer and Activator of Transcription,信号传导和转录激活因子。STAT是一类由细胞因子、生长因子等多肽类配体激活的转录因子,对正常细胞的分化、增殖和凋亡起着重要调控作用。目前在人体中已经发现由不同基因编码的7个成员,分别是STAT1、2、3、4、5A、5B和6。
- 现有研究表明:STAT3存在于大部分肿瘤细胞当中,而在正常细胞中不表达。STAT家族在结构上具有类似性,都含有6个功能区,分别是N端的氨基酸保守区、螺旋区、DNA结合区、连接区、SH2结构域和C端的转录活性区域。STAT家族成员主要通过JAK-STAT信号通路,介导多种细胞因子的下游信号。很多细胞因子和生长因子通过JAK-STAT信号通路传导信号,这些因子包括IL-2、IL-7、CSF、h-GH、EGF、PDGF、IFN等等。这些细胞因子和生长因子在细胞膜上都有相应的受体,这些受体的共同特点是受体本身不具有激酶活性,但受体的细胞内结构域具有酪氨酸激酶JAK的结合位点。
STAT信号通路简介
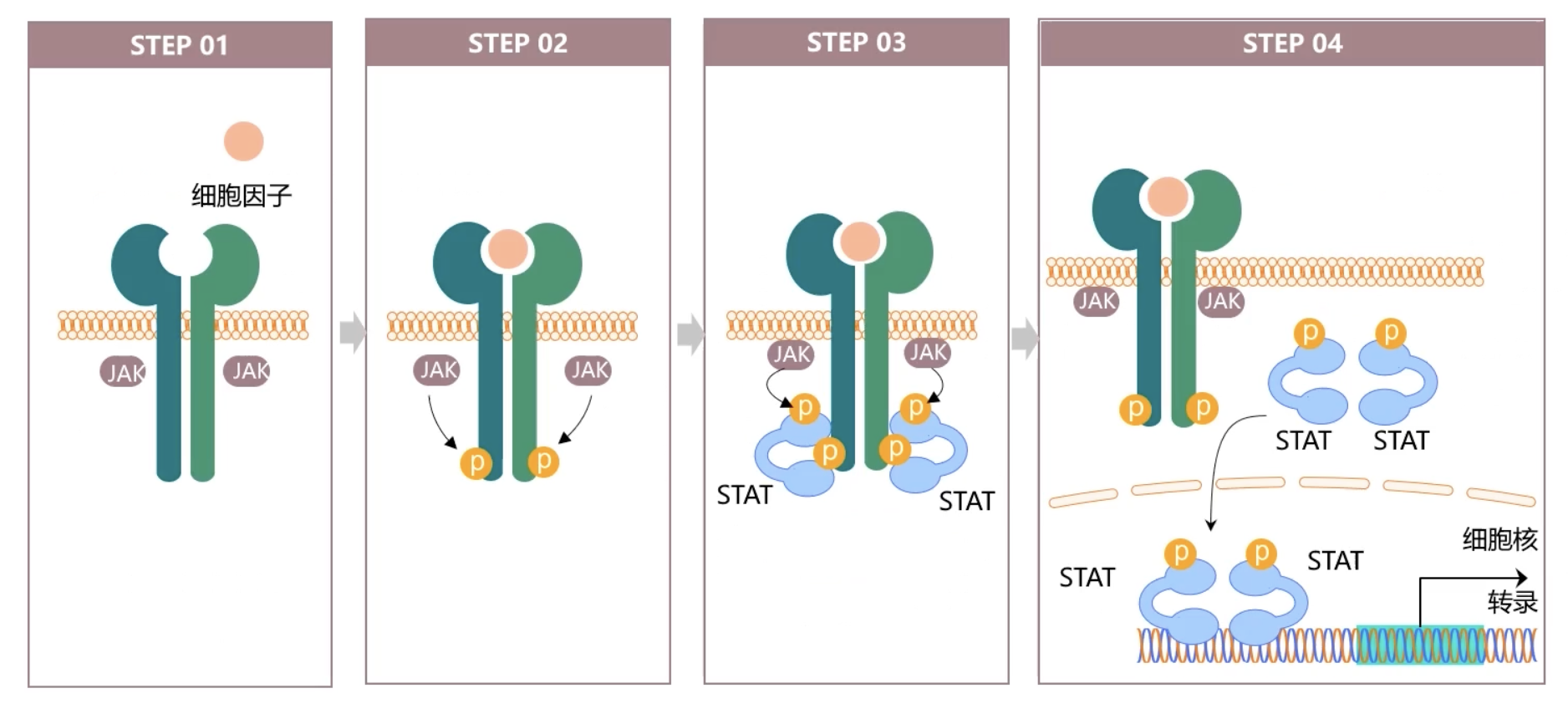
- 细胞因子和相应的受体结合后会引起受体分子二聚体化,受体二聚体化之后使得受体耦联的JAK激酶相互接近并且被活化。
- JAK激活后,催化受体上的酪氨酸残基发生磷酸化修饰,进而这些磷酸化的酪氨酸位点又和周围的氨基酸序列形成一个被称为停泊位点Docking site这样一个特殊结构,吸引含有SH2结构域的STAT蛋白被招募到这个停泊位点之上。
- 最后,激酶JAK催化结合在受体上的STAT蛋白,发生磷酸化修饰,活化的STAT蛋白以同源或者异源二聚体的形式进入细胞核内,与下游的靶基因结合,调控基因的转录。
JAK抑制剂
- 目前已经针对JAK/STAT通路开发了很多的药物,其中不少药物已经获批并且开始了临床应用。在药物的开发思路中,既可以针对JAK开发抑制剂,也可以针对STAT开发相关的抑制剂。
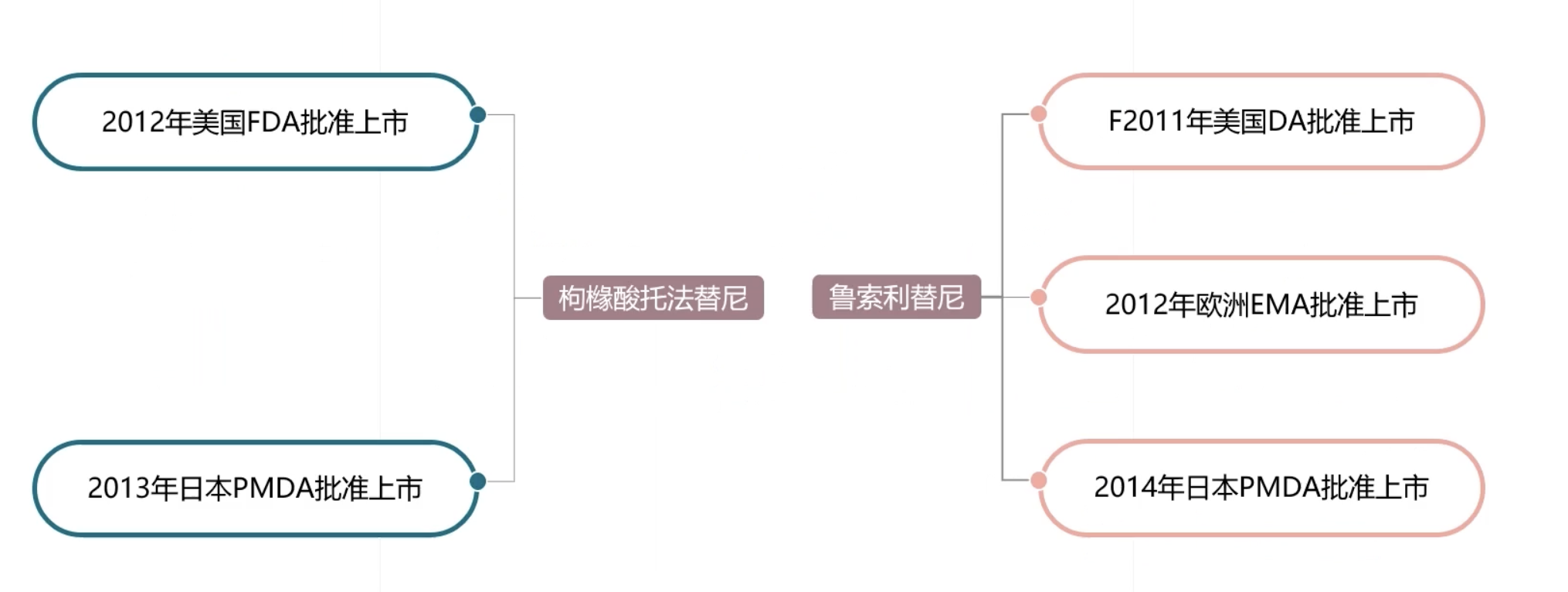
- 在针对JAK的抑制剂当中,很多药物已经获得了批准,例如枸橼酸托法替尼,由辉瑞公司研发,在2012年获得美国FDA批准上市,之后在2013年又获得日本PMDA批准上市。枸橼酸托法替尼是JAK抑制剂,通过干扰JAK/STAT信号通路,从而影响DNA的转录过程。该药物用于治疗成人患者的对甲氨蝶呤应答不充分或不耐受的中度至重度活动性内风湿性关节炎。
- 除此之外,还有鲁索利替尼,由Insight和联合诺华开发。它是JAK1以及JAK2的抑制剂,是第一个获准的专门治疗骨髓纤维化的药物。它在2011年获得美国FDA批准上市,2012年获得EMA欧洲药品管理局批准,在2014年获得日本PMDA批准上市。
STAT抑制剂
- 除了开发激酶JAK的的抑制剂之外,直接抑制STAT活性的抑制剂也同样受到广泛的关注。目前针对STAT的抑制剂共分为三种。
- 第一种是和STAT的DNA结合区域相互结合的抑制剂。目前发现一类新型铂类化合物NSC295558可以特异性地和STAT3的DNA结合区域结合,抑制STAT3的信号通路,从而杀死多种肿瘤细胞株,比如人乳腺癌细胞、胰腺癌细胞、前列腺癌细胞株等,对于STAT5以及STAT1没有抑制作用。NSC295558与STAT3的DNA结合抑制属于非竞争性动力学,可以诱导细胞在G0/G7发生生长停滞和发生凋亡现象。
- 第二种化合物是和STAT的SH2区域结合的抑制剂。化合物NSC74859是一种抑制STAT3活性的小分子化合物。它来自于美国国家癌症研究院化合物库,是通过和STAT3的分子模型比对获得的。体外研究结果和计算机模拟结果一致,这种抑制剂可以在高表达STAT3的肿瘤细胞内,选择性地抑制肿瘤细胞的生长和引起细胞的凋亡,并且通过和STAT3的SH2区域结合而抑制STAT3调节的多种细胞因子的表达,比如Cyclin D1、Bcl-Xl以及Survivin等等。体内实验也证实了NSC74859可以抑制人乳腺癌细胞生长。
- 另一个和STAT3的SH2区域结合的抑制剂是Static。它是通过高通量筛选获得的STAT3抑制剂,属于噻吩类化合物。它的作用机制是通过抑制磷酸化的酪氨酸Motif以及STAT3的SH2区域结合,从而选择性抑制STAT3的活性、二聚体化以及核转位,引起STAT3依赖性乳腺癌细胞的凋亡。
- 第三类抑制剂是和STAT的N端区域结合的抑制剂。STAT3的N端序列由130个氨基酸组成,共有8个螺旋,在基因转录过程中介导了蛋白与蛋白之间的相互作用。因此一种可行的思路是设计短肽类物质与STAT3结合,从而只抑制STAT3的转录活性,而不影响STAT3的磷酸化程度。已经有研究团队通过人工合成的方式合成了两个螺旋结构的类似物,可以和STAT3的N-末端结合。这个人工合成的肽段可以在较低的浓度下特异性诱导人乳腺癌细胞株发生细胞凋亡现象。机制就是通过和STAT3的N-末端结合,阻断STAT3和其他转录调控因子的相互结合和相互作用,从而抑制STAT3借导的下游基因的转录过程。
总结
- 可以看到,针对STAT开发的抑制剂主要都是针对STAT3分子,因为研究表明STAT3存在于大部分肿瘤细胞当中,而在正常细胞中不表达,因此STAT3是一个比较理想的药物开发靶点。
以转录因子为药物靶点存在的问题
- 虽然开发具有转录因子抑制活性的化合物已经有了很多报道,而且有些抑制剂已经开始了临床实验和临床应用,但是目前针对转录因子而开发的抑制剂,大部分的研究还局限于活性筛选的早期阶段。以转录因子为药物靶点开发抑制剂主要存在以下几个问题:
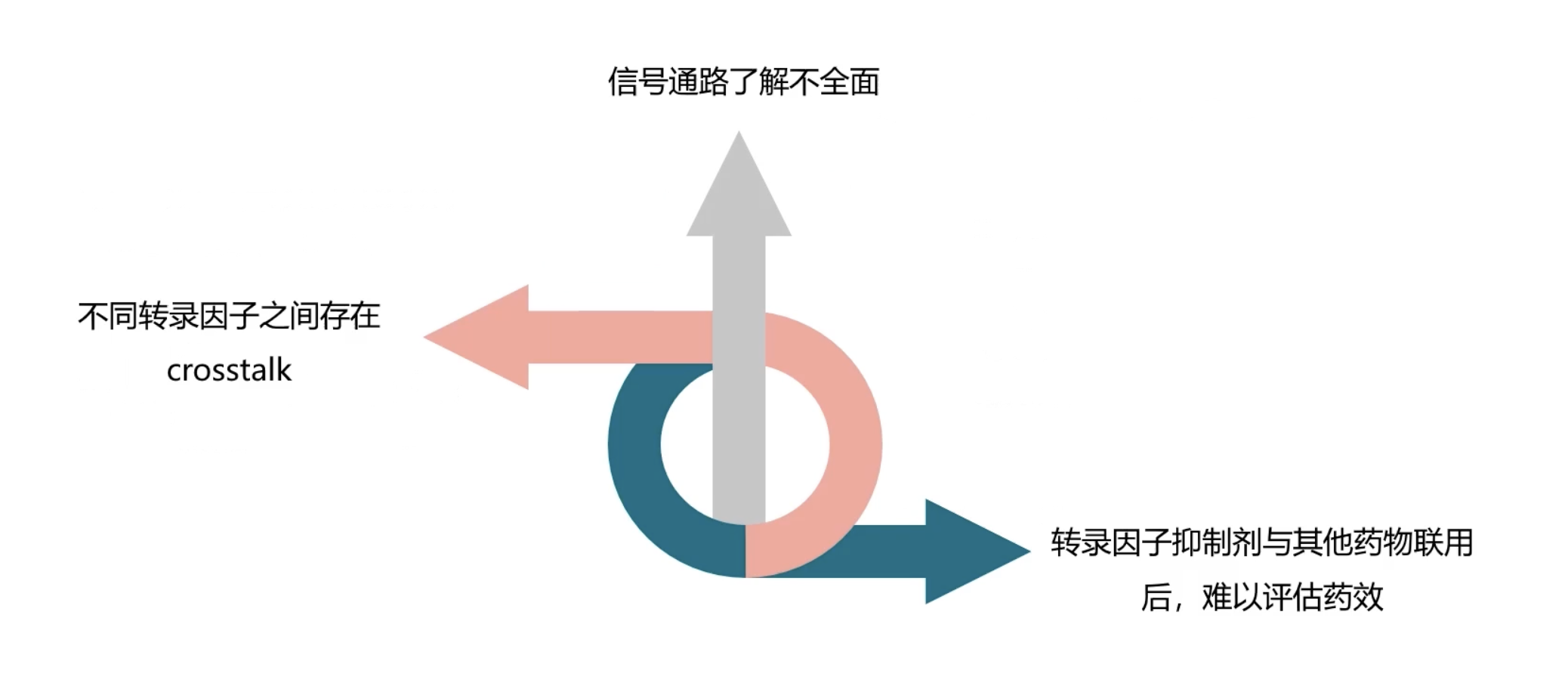
难以获得转录因子在正常生理状态和致癌状态中信号通路的差异。
- 由于转录因子在正常细胞中,特别是细胞的自身免疫系统中,起着重要作用。如果是长期过度地抑制转录因子活性,将不利于正常细胞生长,而产生安全性问题。而且现在抗肿瘤药物都是长期和较大剂量给药,所以目前还很难获得具有瞬间、高度可逆药效的转录因子抑制剂。
很难特异性地抑制转录因子介绍的某个特定的信号通路以及环节。
- 不同转录因子之间存在相互作用和相互交互,也就是cross-talk的问题。它们串联构成一个复杂的信号网络,由于对转录因子在疾病中的作用机制尚未完全阐明,因此无法确定转录因子在肿瘤细胞和正常细胞中是否存在不同的信号通路,或者仅仅只是对同一种细胞因子表达水平的调控的差异。因此难以设计理想的转录因子抑制剂,而特异性地抑制某个转录因子的特定信号通路,而始终保持对其他信号通路没有过度的影响。
难以判断转录因子抑制剂与现有抗肿瘤药物联用后的药效。
- 现有研究表明,抗肿瘤药物的联合用药可以增强疗效并减少不良反应的发生,但由于转录因子本身不具有酶的催化活性,无法特异性地测定它的转录活性或者作用模式,因此转录因子抑制剂和其他抗肿瘤药物的联合用药时,难以直接评估联合用药的药效和疗效。同时,由于转录因子在肿瘤发生中的复杂作用机制,因此如何选择剂型,如何选择给药途径,以及选择和哪种抗肿瘤药物联用等等问题,更是难以进行理性的设计。因此,在未来的道路上还需要花费更大的时间进行临床相关的研究。


