


TIP
这些是本篇文章的标签,来发现更多感兴趣的内容吧
科研
解螺旋
套路课
转录因子
CRISPR-Cas

- 这项技术不算转录因子研究的必要技术,但是因为示例的文章中出现了,还是介绍一下。
实验原理
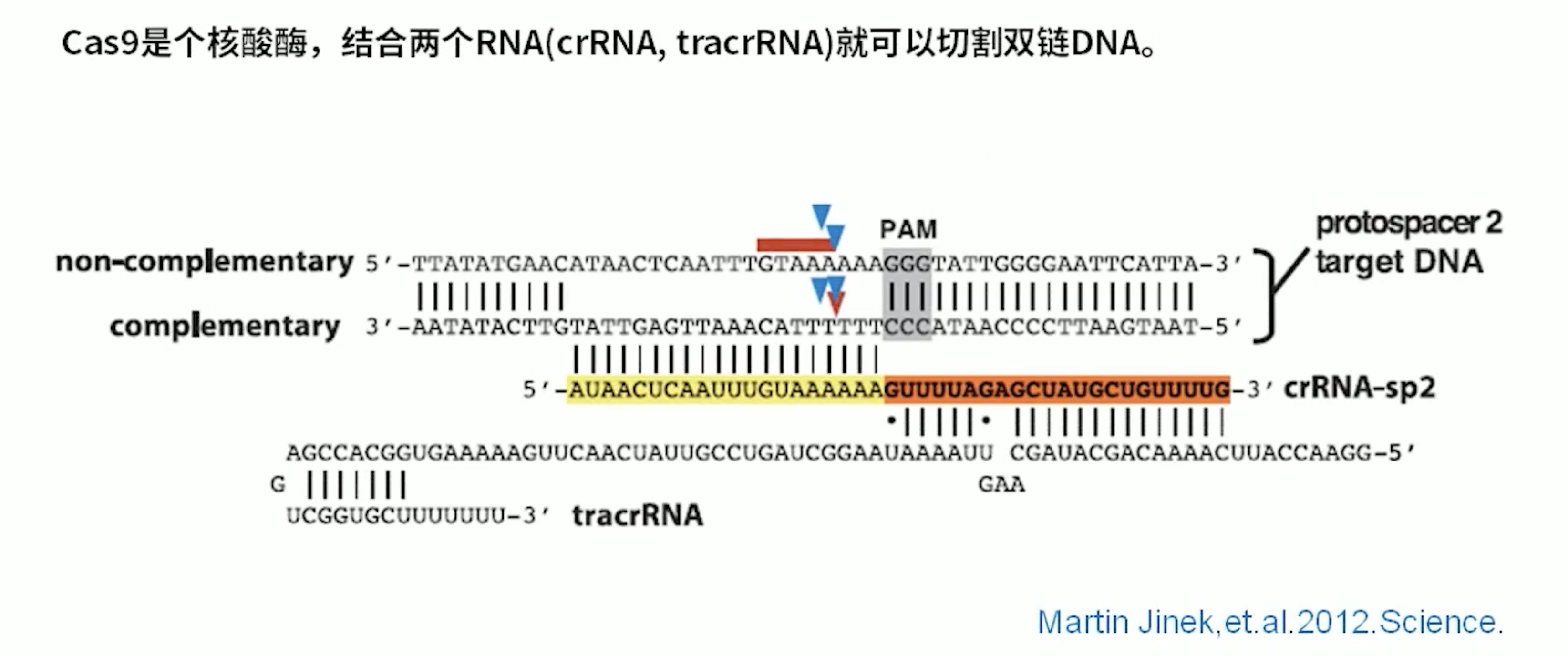
- Cas9它是核酸酶它可以结合两个RNA,分别是crRNA和transRNA,就可以去切割双链DNA。Cas9对序列的特异性切割主要依赖于crRNA与Cas9蛋白形成的RNA蛋白复合物,来识别把序列上的PAM以及Protospacer(序列特点见上),transRNA它对于靶点的识别和切割也是必须的。transRNA的5’-端与成熟的crRNA的3’-端大概有13bp是能够互补配对。这样的结构可能对维持crRNA与靶点的配对是十分重要的。我们将crRNA和transRNA连接起来成为guide RNA。这样可以只需要Cas9蛋白和一条特异的RNA可以进行编辑特异性的DNA。
- Cas9有两个活性位点,这两个核酸酶活性位点分别切割靶DNA的两条链,其中HNH结构域去切割互补的DNA链,RUVC结构域切割非互补的DNA链,所以CRISPR Cas9系统它是可以去切割DNA的双链。
CRISPR-Cas系统的工作流程
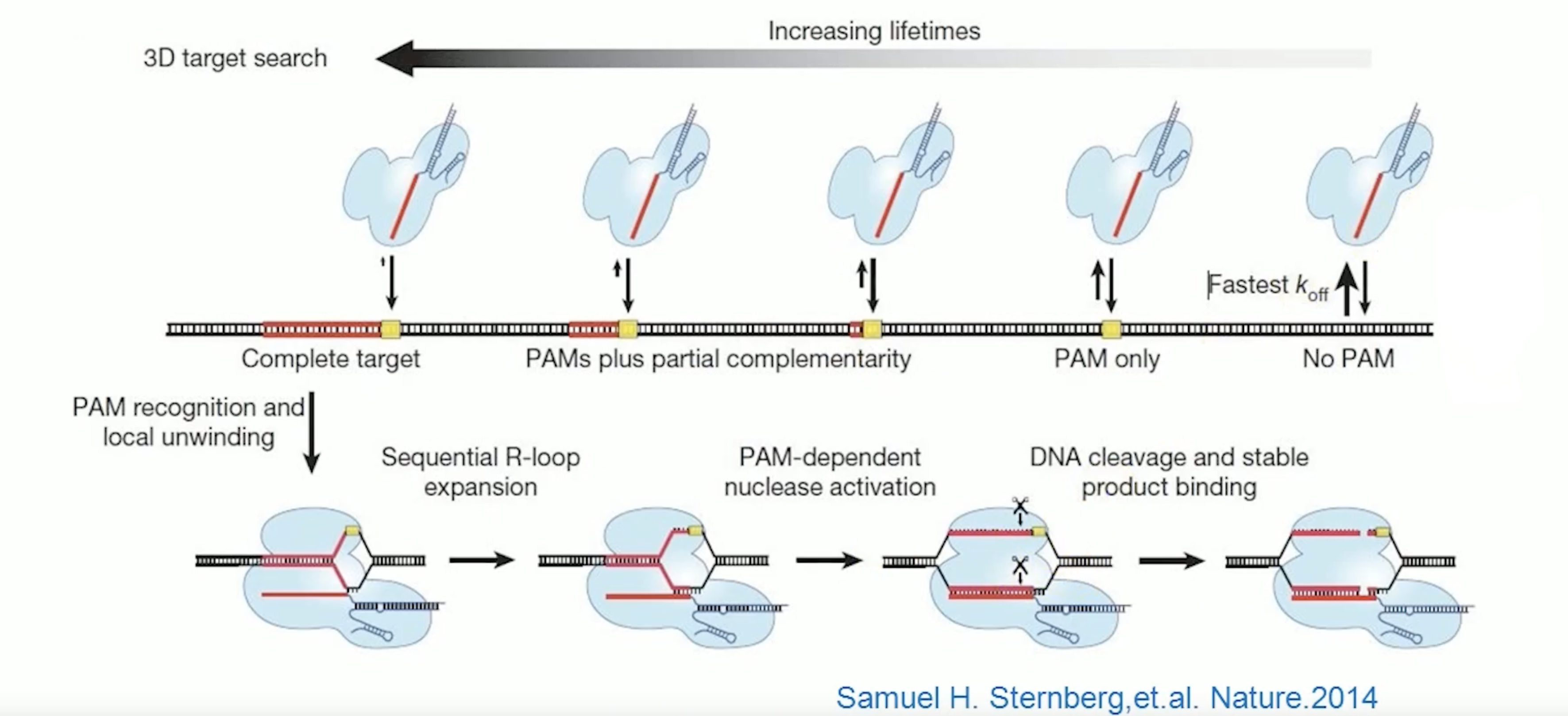
- 我们从右向左看,首先Cas9-RNA复合物去识别PAM序列,如果没有PAM序列,复合物就会迅速解离,从DNA序列上掉下来;识别到PAM序列之后会沿着DNA序列进行搜索,寻找guide RNA与系统互补配对的DNA。
- 当到达正确的位置的时候,Cas9-RNA复合物会使DNA双链打开,形成泡泡样的结构。DNA双链打开之后,RNA可以插入到DNA双链之间,形成RNA-DNA异源双链分子。之后DNA就会被切割了,而且是被切两刀,上面切一刀和下面切一刀。Cas9蛋白有两个活性中心,可以分别切割这两条DNA链。
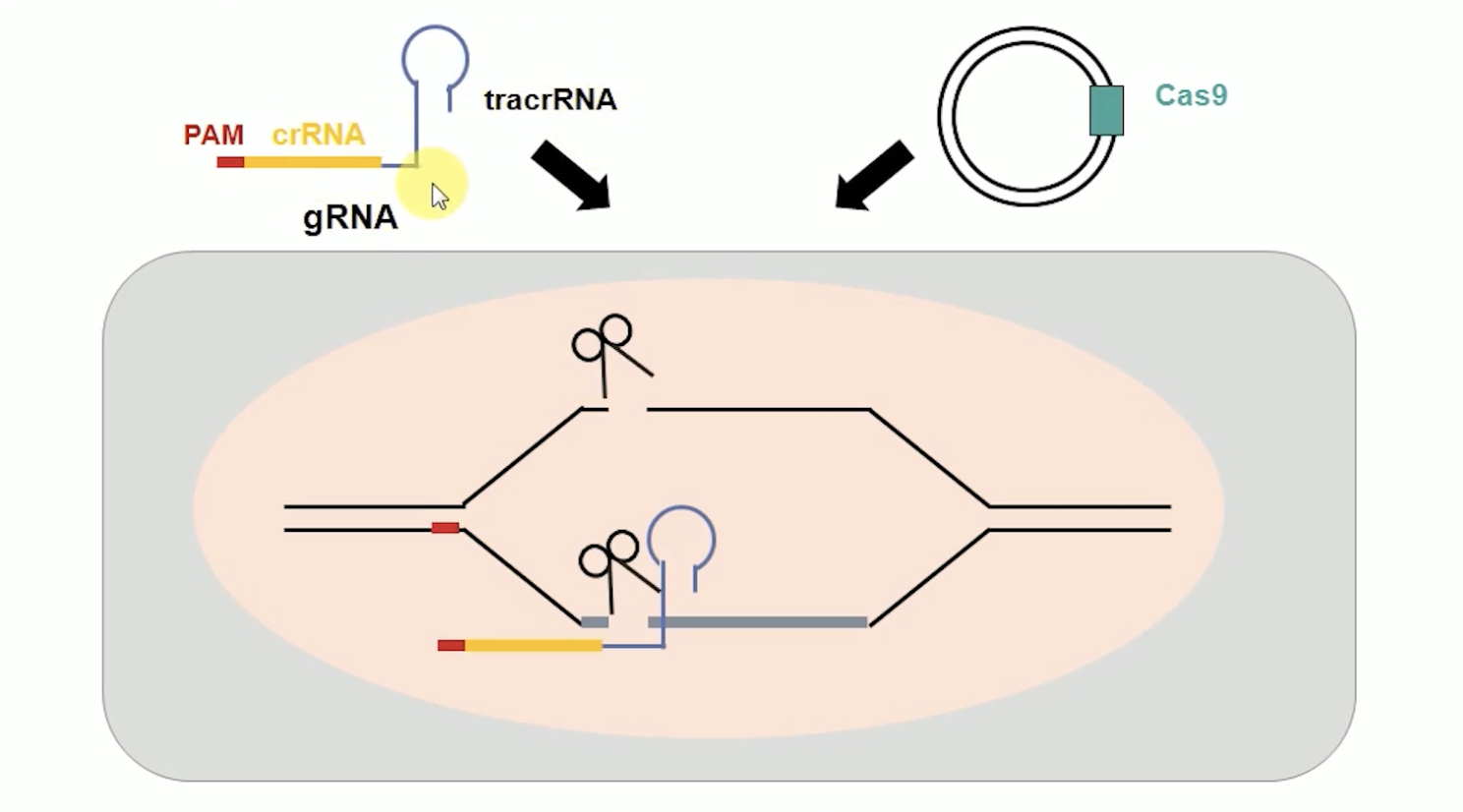
- 首先,待编辑的DNA的区域附近需要相对保守的PAM序列。PAM序列是以NGG为结尾的一段序列,我们还要有guide RNA(guide RNA包括crRNA包括tracrRNA)和Cas9酶的过表达载体。我们将guide RNA和Cas9酶的过表载体给它去共转细胞。
- 当guide RNA能识别到PAM,就能形成RNA-DNA的异源双链;Cas9蛋白就可以去行使切割DNA双链的功能。Cas9可以在基因组DNA上造成缺口,细胞的DNA的修复机制可以将DNA双链上的缺口进行修复;修复的途径有两种,分别是非同源末端连接途径和同源重组的修复途径。同源重组修复途径可以精确得编辑基因组,但是只有在修复模板存在的时候这条途径才能够被启动。人为提供的修复模板是单链的,并不能随机的插入到基因组中的任何位置,而是能够靶向插入到我们设计好的位置,而且这段定向引入的序列能够通过使用PCR技术被迅速高效得检测到。我们能使用PCR来便捷有效的鉴定发生基因组编辑的细胞克隆。
- 利用CRISPR-Cas9进行基因组敲除经常会出现非同源末端连接的现象,这也是高效的进行基因组编辑的重要途径。非同源末端连接是真核细胞中为了避免DNA断裂的累积,强行将两个DNA断端连接到一起的DNA修复机制。在过程中不存在同源序列,因此启动的DNA修复过程不是高保证的,而是会随机的插入基因,随机插入的碱基数目是不一定的,可以是1-200个碱基之间的任何数字。这个途径的优势在于它能造成开放阅读框ORF的改变,导致蛋白翻译的错误,这样蛋白的功能是不完整的这样,以达到基因敲除的目的。
- 这条途径的弊端也是非常明白是,它所造成的碱基插入的数目和位置都是不确定的,鉴定会比同源序修复要麻烦一些。
实验流程
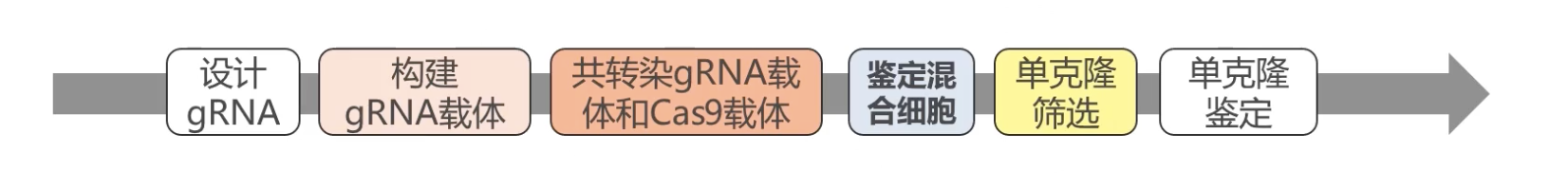
- 首先:设计guide RNA(简写gRNA),注意guide RNA不是随随便便设计出来的,而是要选择位于基因的第一或者第二个外显子的位置(在结构基因的最前面)或者是关键功能结构域,可以使用一些软件去设计,也可以手动设计。
- 第二步:构建guide RNA载体,选择合适的guide RNA载体,将化学合成的guide RNA oligo退火后装入载体。
- 第三步:将装有guide RNA的重组载体和Cas9过表达的载体去共转染目的细胞。
- 第四步:取部分转染后的细胞,抽提基因组DNA,用endo7做酶切鉴定。转染后我们并不知道转进去的东西有没有在乖乖工作,所以要先检查一下,如果检测出来转进去的东西是工作正常的,才能进入下一步。
- 单克隆筛选:一般我们使用的装有guide RNA载体和装有Cas9的过表达载体,这两种载体是带不同的真核抗性基因的,这样可以使用两种药物去给它进行筛选,将没有同时转入两种载体的细胞都杀死。然后使用极限稀释法,克隆环法等等去筛选单克隆。筛选出来的单克隆我们还要再次进行鉴定,因为这种方法造成的缺口,然后造成的剪切插入都是随机的,每个克隆出来的情况都有可能不一样。对挑选的单克隆进行扩大培养,取部分细胞抽取基因组DNA用endo7酶去酶切,同时将PCR产物去送测序鉴定,看看是否造成了基因的移码。
数据分析
- CRISPR-Cas9数据分析的主要内容去鉴定细胞的基因组DNA是否被改变。
- 首先是混合克隆的鉴定,也就是共转染了guide RNA和Cas9过表达质粒之后还没进行单克隆筛选的细胞。在细胞中引入Cas9和guide RNA并不能将所有的细胞都成功编辑,而是将带来混合的细胞群体。双链断裂之后细胞内的修复机制通过非同源末端连接来起作用,非同源末端连接会带来插入缺失的错误。不过引入的插入缺失它是存在抑制性的,而等位基因编辑的频率也有所不同。有的细胞没有被编辑,有的只有一个等位基因被编辑,而有的两个等位基因都被编辑了。可能这种杂合的或者是没有被编辑的情况会比较多一,点验证过程的第一步是要快速评估是否有一定量的细胞被编辑了,对于插入缺失而言可以通过错配切割分析来观察。
- 混合克隆中比较多见的情况是野生型和杂合型。我们可以把混合克隆的基因组DNA抽提纯化出来,以它为PCR模板进行PCR扩增,把我们的目标区DNA用PCR大量扩增出来。我们做PCR扩增不仅要扩增编辑的DNA片段,同时要取没有做任何处理的野生型细胞去抽基因组DNA,然后扩增成为野生型的DNA片段,为下一步做酶切分析准备。
- 所以之前我们抽基因组DNA的时候,在抽提转染了guide RNA和Cas9载体的细胞的基因组DNA的同时,也应该要抽提没有经过转染的野生型细胞的基因组DNA。把基因组编辑和野生型组的基因组DNA上的目标片段扩增出来后,我们可以用PCR产物回收试剂盒,把PCR产物给纯化了,测量好浓度,进入下一步的酶切分析。
- 酶切分析,也叫做错配切割分析,通常是包含两个步骤,首先是变性DNA,并且让转染组的DNA和野生型的DNA链退火杂交,接着要用酶去孵育退火的DNA,以切割异源双链。第三,进行琼脂糖凝胶电泳分析。错配切割分析是一种非常快速而简单的插入缺失检测方法,在过程中可以使用很多种核酸酶,这里以T7 Endo为例。
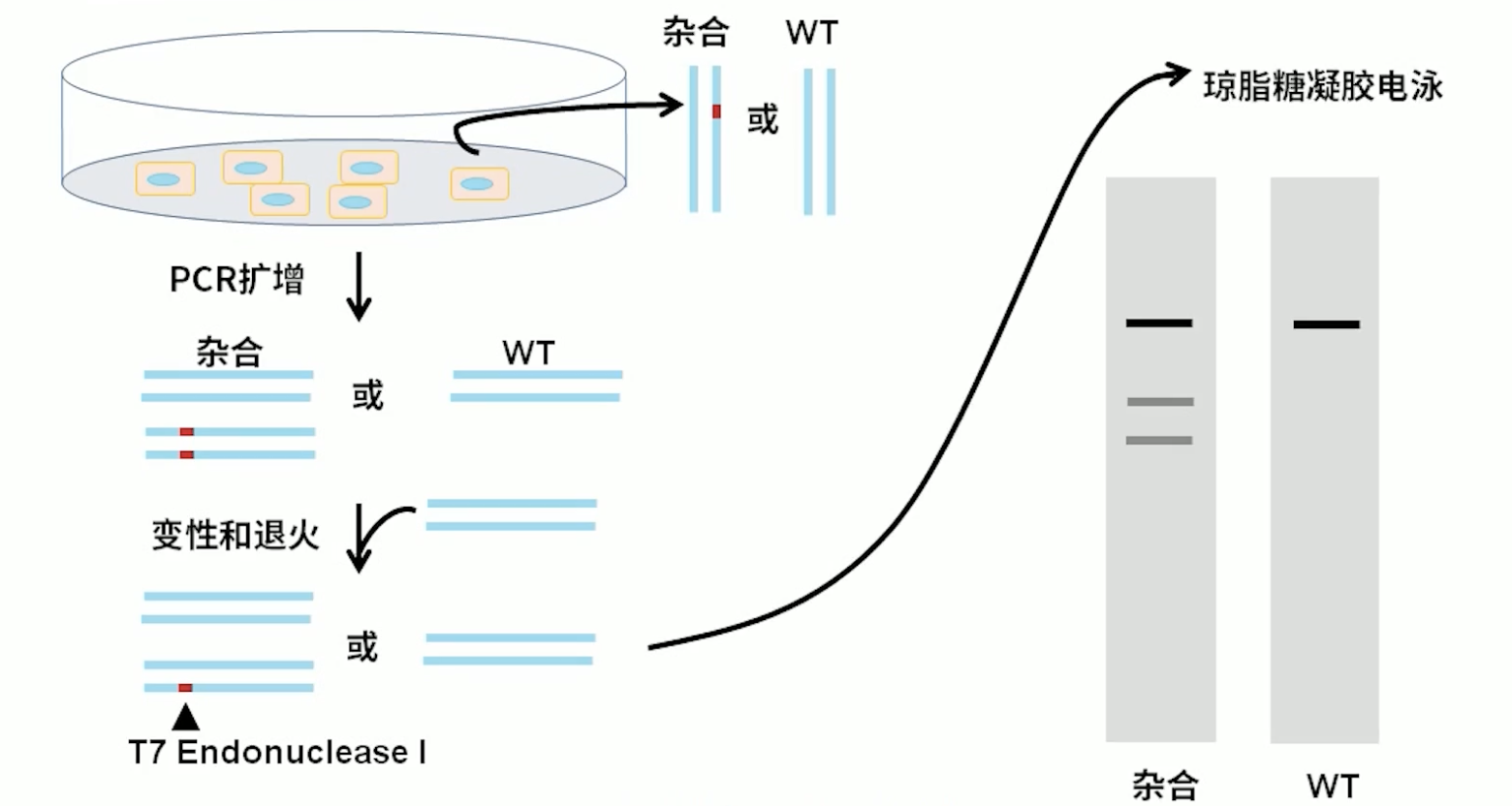
- 前面我们已经准备好PCR产物了,转染组的PCR产物可能有两类,混合细胞中是野生型和基因编辑过的混合物或者完全都是野生型的。我们把野生型细胞的PCR产物和转染组的PCR产物退火之后,杂交组退火出来的产物有两种,一种是DNA双链中有一条链是经过编辑的这种退火产品能够被T7 Endo识别并且切割成两条小的片段;另一种是双链中任何一条链都没有被编辑过,这就不能被切割。跑胶时可以看到三条带。野生型这一组退火出来的产品肯定都是沒经过编辑的,T7 Endo沒办法去识别和切割,跑胶时可以看到一条带。
- 单克隆的鉴定也是用同样的方法,如果我们挑到的单克隆是两条等位基因都被编辑过的,那经过PCR扩增-变性-退火之后得到的产物就应该是一条链野生、一条链是编辑过的杂合链,都能被T7 Endo识别和酶切,跑胶出来会是两条短一些的条带。
常见问题

- 关于第三点:shRNA技术如果编辑效率足够是可以不筛选单克隆的,也不用构建稳转株。但CRISPR-Cas9基因编辑技术的编辑效率没有那么高,可能只有20-30%。
